文献分享:癌症免疫衰老图谱的多组学与单细胞特征分析
写在前面(博客)
今天在课题组里汇报了一篇文献,后面想想这篇文献其实挺适合作为数据挖掘——多组学——多学科网页工具三部曲的一个典型示范的,遂把当时的汇报文档分享至博客。
本文语言已尽可能生动有趣,因为一开始便打算在汇报时实现哪怕是完全的门外汉也能听懂的效果。
流程:
- 简单介绍文章思路
- 讲解文章的定义方法
重点介绍本文的shiny应用,欣赏一些图片- 一气呵成地说“我的分享到此结束谢谢大家”
Multi-omics and single cell characterization of cancer immunosenescence landscape
Title:癌症免疫衰老图谱的多组学与单细胞特征分析
感觉是个不太切题的题目,应该叫“PCA分析的绝佳应用以及多组学可视化大赏(doge”
07 July 2024 发表于《Scientific Data》
重点产出结果:https://bio-pub.shinyapps.io/cs_nomo/ (但是……)
原文摘要如下:
Cellular senescence (CS) is closely related to tumor progression. However, the studies about CS genes across human cancers have not explored the relationship between cancer senescence signature and telomere length. Additionally, single-cell analyses have not revealed the evolutionary trends of malignant cells and immune cells at the CS level. We defined a CS-associated signature, called “senescence signature”, and found that patients with higher senescence signature had worse prognosis. Higher senescence signature was related to older age, higher genomic instability, longer telomeres, increased lymphocytic infiltration, higher pro-tumor immune infiltrates (Treg cells and MDSCs), and could predict responses to immune checkpoint inhibitor therapy. Single-cell analysis further reveals malignant cells and immune cells share a consistent evolutionary trend at the CS level. MAPK signaling pathway and apoptotic processes may play a key role in CS, and senescence signature may effectively predict sensitivity of MEK1/2 inhibitors, ERK1/2 inhibitors and BCL-2 family inhibitors. We also developed a new CS prediction model of cancer survival and established a portal website to apply this model (https://bio-pub.shinyapps.io/cs_nomo/).
细胞衰老(Cellular Senescence, 简称 CS)与肿瘤进展密切相关。然而,目前针对人类癌症中衰老基因的研究尚未深入探讨癌症衰老特征与端粒长度之间的关系。此外,单细胞分析尚未揭示恶性细胞和免疫细胞在衰老水平上的演化趋势。在本研究中,我们定义了一种衰老相关特征,称为“衰老特征(senescence signature)”,并发现衰老特征得分较高的患者预后较差。衰老特征得分较高与较高的年龄、更高的基因组不稳定性、更长的端粒、淋巴细胞浸润增加、更高的促肿瘤免疫浸润(包括调节性T细胞和髓源性抑制细胞)密切相关,并能预测免疫检查点抑制剂治疗的响应情况。单细胞分析进一步揭示,恶性细胞和免疫细胞在衰老水平上具有一致的演化趋势。MAPK信号通路和凋亡过程可能在衰老中发挥关键作用,而衰老特征可以有效预测对MEK1/2抑制剂、ERK1/2抑制剂和BCL-2家族抑制剂的敏感性。我们还开发了一种新的癌症衰老预测模型,并建立了一个门户网站以应用该模型(https://bio-pub.shinyapps.io/cs_nomo/)。

让我们说中文:
首先,为什么要做
- 细胞衰老(CS)与癌症进展密切相关,对肿瘤微环境有着显著影响
- 然而细胞衰老过程非常复杂,且缺乏单一的特异性标志物
衰老是一种由一系列相关细胞变化组成的综合征,包括端粒变化、染色质重组、基因表达谱、分泌组(secretome)和代谢通路的变化
- 目前缺乏针对“衰老”这一复杂变化的量化比较指标
其次,怎么样去做
- 通过分析细胞衰老基因表达谱定义了一种指数,称为“衰老特征(senescence signature)”
- 基于这一新定义出来的指数,开发了一种新的细胞衰老预测模型,并建立一个shiny网页工具来预测泛癌患者的预后
最后,做了又怎样
通过衰老特征(senescence signature)这一指数,
- 作为预测泛癌症预后的可靠指标
- 显著预测各种癌症的免疫治疗响应
- 与肿瘤的免疫特性、多组学变化、端粒变化密切相关
发现:
- 衰老特征能够反映癌症间及癌症内的异质性,并显著分类患者的生存状况
- 较高的衰老特征得分与泛癌中的更高基因组不稳定性和增殖水平相关
- 较高的衰老特征得分与泛癌中的淋巴细胞浸润增加及更高的促肿瘤免疫浸润相关
- 较高的衰老特征得分与较长的端粒和较高的TERT表达在泛癌中相关
- 衰老特征能够预测免疫治疗和靶向治疗的反应
衰老特征(senescence signature)怎么被捏出来的?
衰老特征的定义
为了筛选与生存相关的衰老基因,我们使用Cox回归模型,通过调整每种癌症类型中的年龄和临床分期,获得每个CS相关基因的HR值。我们将HR小于1且p值小于0.05的基因(在至少5种癌症类型中)定义为保护因子,将HR大于1且p值小于0.05的基因定义为风险因子。筛选后,保留了68个基因(表S4)。然后,我们使用这些基因为每个聚类定义衰老特征。衰老特征通过以下两个步骤定义:首先,通过PCA分析获得每个基因的PC1得分;其次,将PC1得分乘以相应的基因表达值,并将结果相加:
senescence signature = Σ(PC1 × G_i)
其中,i 是68个潜在特征基因之一,G 是该基因的表达值。根据衰老特征得分的百分位数,我们将肿瘤样本分为5组:group1(0–20%)、group2(20%–40%)、group3(40%–60%)、group4(60%–80%)、group5(80%–100%)。

为了方便理解,做了一份Mermaid流程图
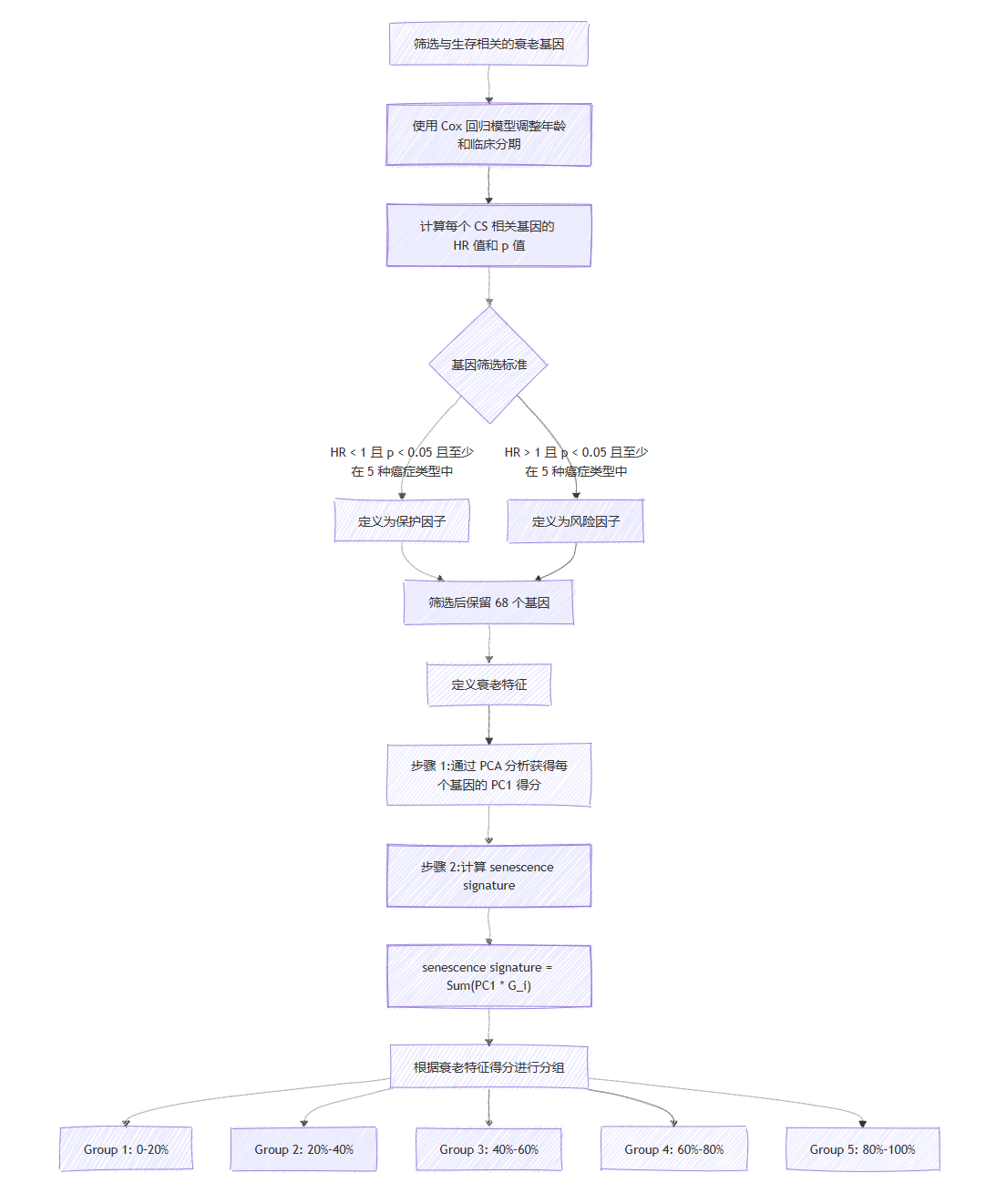

道理我都懂,但是你在说什么?

这个流程是比较清楚了,但是不少细节完全不明白
CS相关基因怎么得到的?
再来简化一下流程图
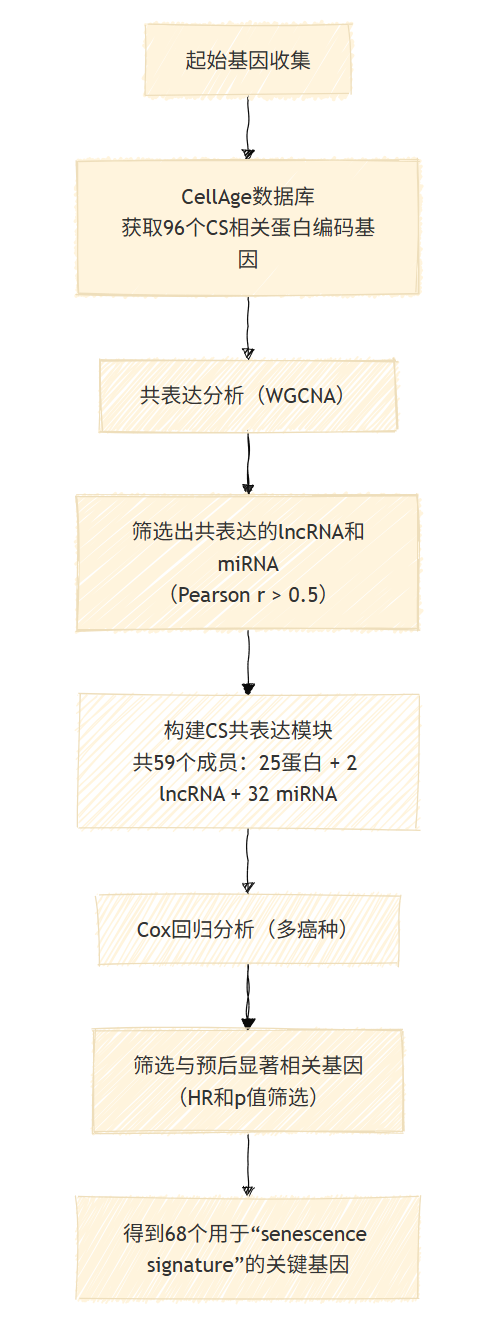
- 通过加权基因共表达网络分析(WGCNA),鉴定出一个与CS高度相关(皮尔逊相关系数r>0.5)的基因共表达模块,称为CS模式(CS patterns)。该模块包括25个CS相关的蛋白编码基因、2个lncRNA以及32个miRNA(图S1A)
首先筛选出与CS相关的蛋白编码基因、lncRNAs和miRNAs,这些分子与CS基因之间的Pearson相关系数大于0.5,被认为具有共表达关系;
然后使用WGCNA(v1.69)构建共表达网络
我们可以看看使用Cytoscape进行可视化的效果
PCA分析又是怎么进行的
在本文中,主成分分析(PCA) 被用来检验基于59个CS相关基因(WGCNA模块中的25蛋白基因 + 2 lncRNA + 32 miRNA)是否能够区分肿瘤组织和配对的正常组织。
样本选择:
- 从TCGA中筛选出包含不少于3对肿瘤-正常配对样本的癌种(最终选出14种癌症)。
数据准备:
对这59个CS相关基因的表达量分别取 log2 转换后求平均:

PCA分析执行:
- 对每个样本在59个基因上的表达做PCA降维。
- 提取前两个主成分 PC1 和 PC2,用于二维可视化,查看是否能在空间上区分肿瘤与正常组织。
PCA结果评估:
- 以PCA得分(score = PC1 + PC2)作为判别指标。
- 用ROC曲线评估PCA得分在区分肿瘤与正常组织方面的性能。
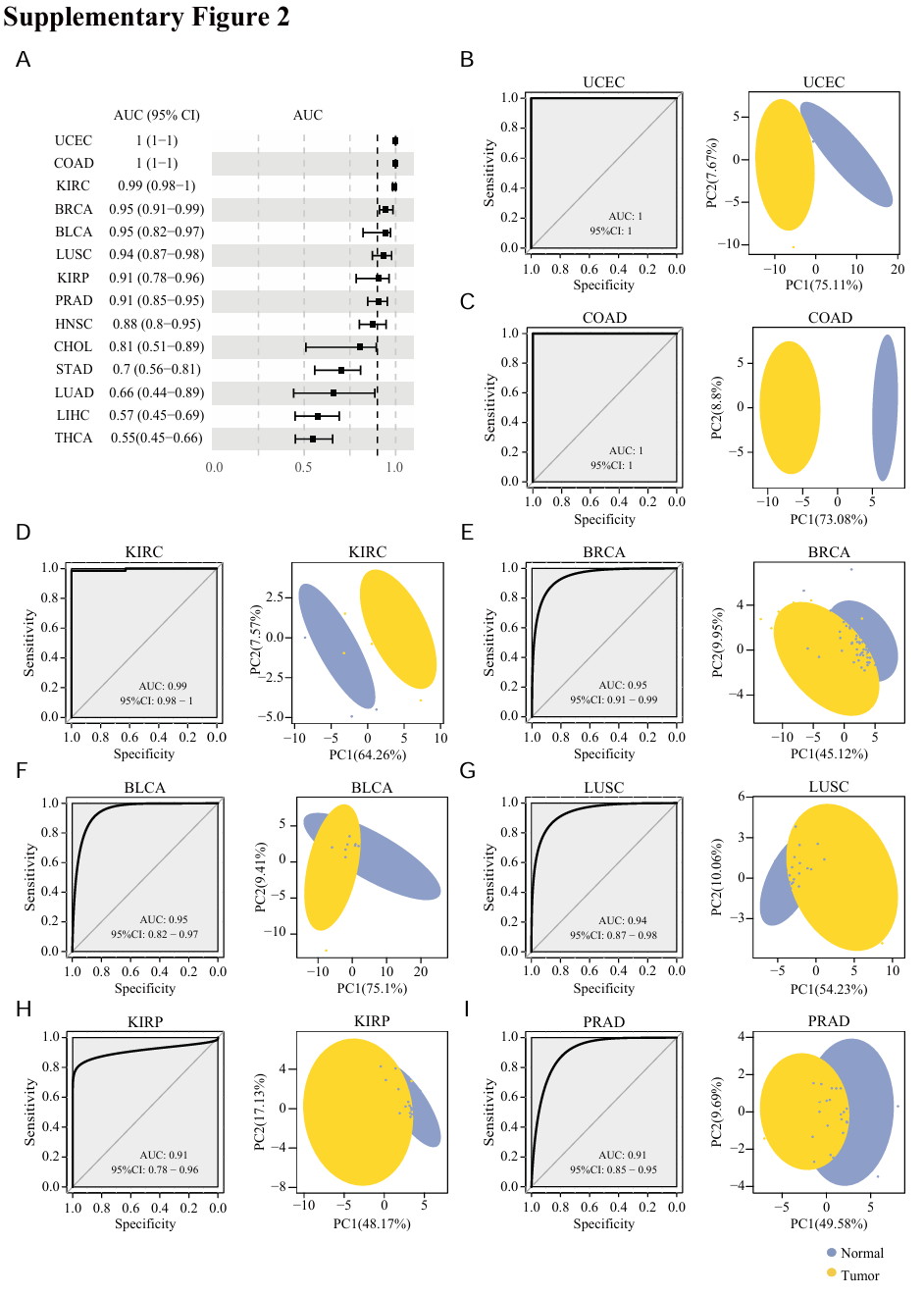
- 结果显示 9种癌症的AUC > 0.9,表现出非常高的判别能力。

回到熟悉的shiny!
这是一个很典型的反面案例,作者似乎并没有及时维护以及预存数据(不知道是不是中美关系变化的缘故),好早前就联系了通讯作者反馈了情况但是至今没有任何答复,所以这个网站
完——全——用——不——了!

不过思路已经是很通畅的了,大家自己看看示例图意会一下吧

这幅图是使用说明中配的,但是存在很诡异的地方,仔细观察可以发现ACC和UVM的预期生存曲线是完全一样的,不管男性女性都没有任何区别……肾上腺皮质癌和原发性眼内癌完全没什么关联性啊
或许一开始,这个网页工具就存在不少问题了,现在则是彻底的罢工……
引以为戒
我来当当艺术展讲解员~
全文总计有80张,10余种不同类型的图像。可谓是视觉盛宴,让人眼花缭乱

Circos plot

但是这居然是使用Cytoscape (v3.9.1)绘制的,有点出乎意料
网络图
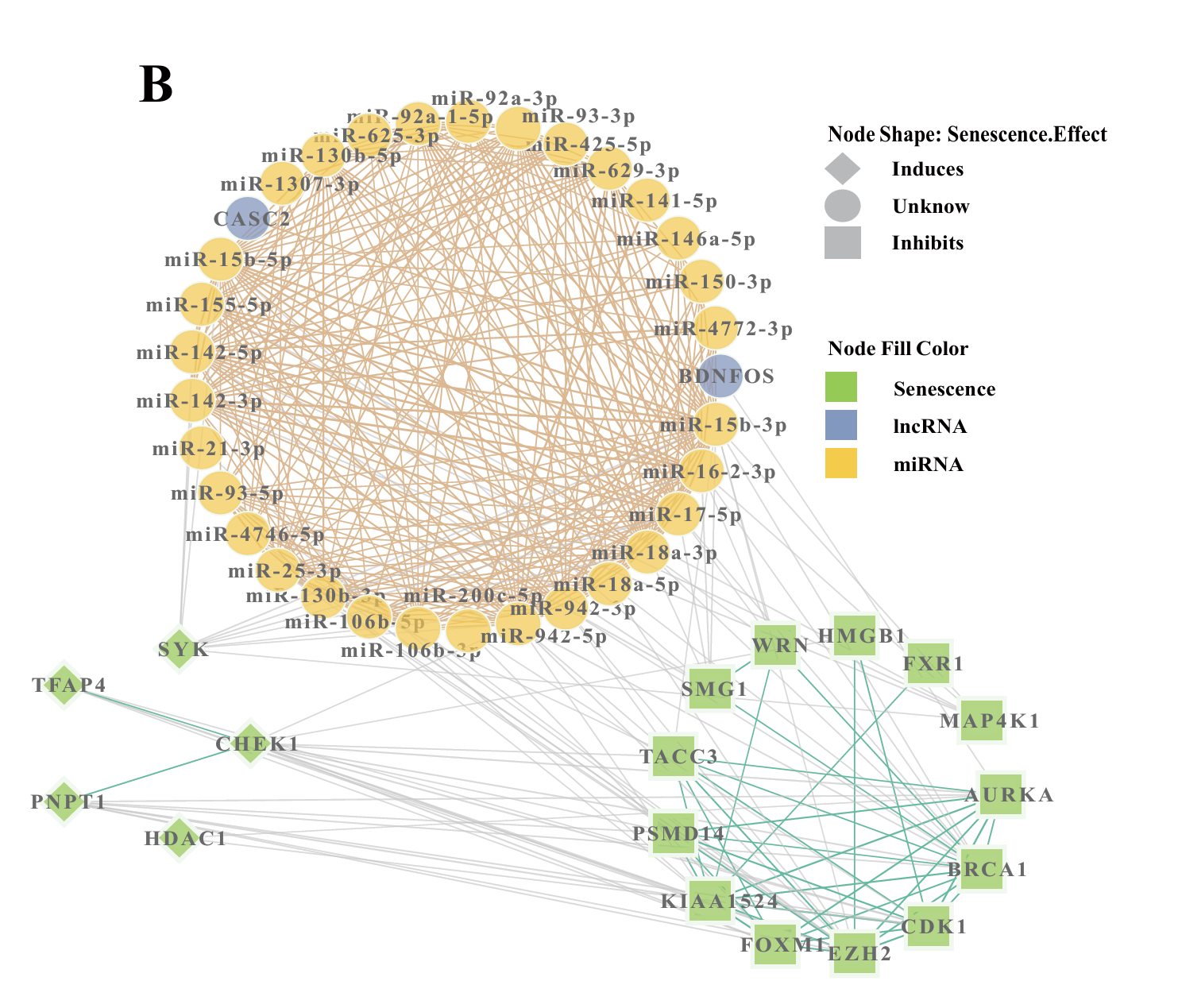
使用Cytoscape (v3.9.1)绘制
PCA图+ROC曲线

关于数据来源
- TCGA(https://portal.gdc.cancer.gov/):33种癌症类型中10,510个样本的临床信息、mRNA、miRNA和突变数据集
- CellAge数据库(http://genomics.senescence.info/cells):与CS相关的蛋白编码基因
- DepMap数据库(https://depmap.org/portal/):获取CRISPR基因组筛选数据集
- GDSC(https://www.cancerrxgene.org/):不同细胞系中不同药物的IC50数据
关于工具与R包使用
| R 包名 | 版本 | 主要用途 |
|---|---|---|
| WGCNA | v1.69 | 加权共表达网络分析(构建CS模块) |
| survival | v3.2-13 | 生存分析,Cox回归,Kaplan-Meier曲线 |
| survminer | v0.4.9 | 美化生存分析图表(KM曲线等) |
| limma | v3.46.0 | 差异表达分析(miRNA、蛋白表达) |
| ssGSEA | v1.38.2 | 单样本基因集富集分析(免疫浸润评分) |
| scanpy | v1.9.2 (这个是Python用的) | 单细胞转录组分析(注意:这是Python的) |
| Harmony | 作者没写,我也不知道 | 批次效应校正(单细胞整合) |
| Seurat | v4.1.0 | 单细胞数据处理和可视化(细胞类型注释) |
以及以下工具:
- Cytoscape v3.9.1:可视化共表达网络、绘制Circos plot;
- CellPhoneDB(v4.0.0):细胞间通讯分析(Python环境)
感想
至此,这篇文献的最重要且主要的内容就基本讲完了。
这一篇文献运用了大量可视化以及算法来进行研究,其研究思路抽丝剥茧步步为营,值得我们学习,每一份图都是经典的运用,我们时间有限不能一一详细论述,大家感兴趣可以来“观摩一下”原文,它的附加图片比正文图片更加精彩!
最后,用思维树来全面概括一下这篇文章

分享结束,谢谢大家

最后,祝大家代码不报错,数据超纯净,运行一遍过

本文献所有图片整理
文献正文(Figure 1A–6F)
36+44=80张图
| 图号 | 图像类型 | 图像内容及作用 | 图像解读方法 | 图像绘制方法 |
|---|---|---|---|---|
| Figure 1A | 条形图 | 展示33个癌种中senescence signature的中位值分布 | 观察不同癌种中senescence程度高低 | ggplot2 |
| Figure 1B | 箱线图 | 比较CS clusters 1-3之间senescence signature的差异 | signature得分越高,表明衰老水平越高 | ggplot2 + Wilcoxon检验 |
| Figure 1C | 箱线图 | 比较不同senescence分组之间的年龄差异 | 验证signature与年龄的正相关性 | ggplot2 |
| Figure 1D | Kaplan-Meier生存曲线 | 比较3个CS clusters在整体生存(OS)上的预后差异 | cluster 3 生存最差,cluster 1 最好 | survminer |
| Figure 1E | Kaplan-Meier生存曲线 | 比较5个senescence score分组在OS上的生存差异 | senescence得分越高,预后越差 | survminer |
| Figure 1F | 模型流程图(网页截图) | 展示CS预测模型的在线使用平台 | 概览预测模型结构与应用界面 | 图形设计工具(Figdraw等) |
| Figure 2A-B | 热图 | 展示SNV和CNV在高低CS组中的显著差异 | 颜色代表突变或CNV频率,观察显著通路 | ComplexHeatmap |
| Figure 2C-E | 箱线图 | 比较CS clusters之间基因组不稳定性指标(断点、异倍性、异质性) | signature越高,基因组不稳定性越强 | ggplot2 + Kruskal-Wallis |
| Figure 2F | 条形图 | 展示高低CS组在关键信号通路突变频率的差异 | 如TP53, MAPK等通路在高组中频率更高 | ggplot2 |
| Figure 2G-H | 条形图(表达差异) | 展示高低CS组中差异表达的miRNA和蛋白 | 区分上调和下调,通路颜色编码 | limma + ggplot2 |
| Figure 3A | 箱线图 | 展示不同CS组别中28种免疫细胞的浸润分值 | 观察高CS组免疫浸润水平是否升高,尤其是Treg和MDSCs | ssGSEA计算免疫浸润 + ggplot2 |
| Figure 3B | 条形图 | 展示不同CS clusters中各免疫细胞浸润水平的趋势 | 免疫细胞组成随CS signature升高发生变化 | ggplot2 |
| Figure 3C | 条形图 | 免疫通路基因表达在不同CS组别中的差异 | 观察共刺激、共抑制通路是否活跃 | ssGSEA + ggplot2 |
| Figure 4A-E | 箱线图 | 比较高低CS组在端粒长度、TERT表达、TVR等方面的差异 | 高CS组通常端粒更长,TERT表达升高 | Wilcoxon检验 + ggplot2 |
| Figure 4F-H | 条形图/箱线图 | 展示shelterin基因的表达在CS组间的变化 | 识别CS signature对端粒调控蛋白的调节 | ggplot2 |
| Figure 5A-B | UMAP | 展示539350个单细胞及其细胞类型分类 | 识别恶性细胞、免疫细胞分布,辅助后续分析 | Seurat/scanpy |
| Figure 5C-D | 相关散点图 | CS signature在免疫与肿瘤细胞之间的相关性(单细胞平均) | 皮尔逊相关性,值越高说明共演化越一致 | ggplot2 |
| Figure 5E | 箱线图 | 展示药物处理后GBM细胞中CS signature的变化 | 药物干预是否降低senescence水平 | ggplot2 |
| Figure 5F-G | 互作强度条形图/热图 | 分析高低CS组肿瘤-免疫细胞互作强度与受体配体对差异 | 颜色/数值代表互作强度,CellPhoneDB输出 | CellPhoneDB + ggplot2 |
| Figure 6A-D | 箱线图 + 生存曲线 | 比较免疫治疗响应患者的CS signature以及其对生存的预测能力 | 响应者CS signature较低,生存更好 | ggplot2 + survminer |
| Figure 6E-F | 散点图(相关性) | CS signature与靶向药物IC50之间的相关性分析 | 相关系数越高表示预测能力越强 | Spearman相关分析 + ggplot2 |
补充材料(Supplementary Figure 1A–18)
| 图号 | 图像类型 | 图像内容及作用 | 图像解读方法 | 图像绘制方法 |
|---|---|---|---|---|
| Supplementary Figure 1A | Circos图 | 展示CS相关基因在全基因组的染色体分布与共表达关系 | 观察哪些基因对共表达频繁,以及miRNA是否为关键连接者 | Cytoscape(圆形布局模拟Circos) |
| Supplementary Figure 1B | 网络图 | 可视化CS相关基因(lncRNA/miRNA/蛋白编码基因)之间的共表达调控网络 | 节点颜色代表类型,形状代表功能(诱导/抑制/未知),连线为r>0.5的共表达关系 | Cytoscape |
| Supplementary Figure 1C | 热图(Heatmap) | 展示CS基因在16种癌症中肿瘤组织与正常组织的表达差异 | 红色代表上调,蓝色代表下调,重点关注CS抑制基因的上调趋势 | pheatmap 或 ComplexHeatmap |
| Supplementary Figure 1D | ROC曲线 + PCA图 | 展示CS signature在区分肿瘤与正常组织上的分类性能(AUC值) | AUC>0.85表示优秀,PCA图中两组分离度越高效果越好 | pROC + ggplot2 |
| Supplementary Figure 2A | 条形图 | 显示14种癌种中使用CS signature区分肿瘤与正常的AUC评分 | 越接近1表示分类效果越好 | ggplot2 |
| Supplementary Figure 2B–I | PCA图 + ROC曲线 | 展示8种癌症中CS signature在PCA维度与分类效能上的表现 | 观察是否肿瘤与正常可被第一主成分清晰分离,及其AUC | prcomp + ggplot2 + pROC |
| Supplementary Figure 3A–F | PCA图 + ROC曲线 | 补充其余6种癌症中CS signature的分类效能表现 | 与Figure 2B–I相同,识别诊断性强的癌种 | prcomp + ggplot2 + pROC |
| Supplementary Figure 4 | 热图(Heatmap) | 展示33种癌症中CS相关基因的体细胞突变频率和类型 | 颜色越深表示突变频率越高,查看突变类型分布 | ComplexHeatmap |
| Supplementary Figure 5A | 折线图(elbow plot) | 展示K-means聚类的最佳聚类数为3 | 寻找拐点位置作为聚类数参考 | factoextra::fviz_nbclust |
| Supplementary Figure 5B | 条形图 | 展示CS clusters在各个癌症类型中的分布数量 | 分析哪类癌种更集中于某个cluster | ggplot2 |
| Supplementary Figure 6A–C | 森林图(Forest plot) | 展示CS clusters对OS, DSS, PFI三种生存指标的风险预测能力 | HR>1表示风险增加,关注95%CI是否跨1 | forestplot 或 ggforest |
| Supplementary Figure 7A | 箱线图 | 比较不同CS clusters之间的患者年龄差异 | 趋势性分析,cluster 1 年龄最小 | ggplot2 + Kruskal-Wallis |
| Supplementary Figure 7B | 堆积条形图 | 展示AJCC临床分期在CS clusters中的分布情况 | CS越高,晚期患者比例越高 | ggplot2 |
| Supplementary Figure 8A–B | 箱线图 | 展示CS clusters中增殖水平、突变率和新抗原负荷的变化 | 观察是否随CS分组递增,呈现正/负相关 | ggplot2 + Kruskal-Wallis |
| Supplementary Figure 9A | 箱线图 | 比较不同CS组别中的免疫和基质分数 | 分数升高说明TME活跃度上升 | ESTIMATE + ggplot2 |
| Supplementary Figure 9B | 气泡图(bubble plot) | CS高组上调基因的KEGG通路富集结果 | 气泡大小代表富集基因数,颜色代表p值 | clusterProfiler + ggplot2 |
| Supplementary Figure 10 | 热图 + 条形图 | 展示高低CS组在多个癌种中免疫-肿瘤细胞之间的配体-受体相互作用 | 比较高低组互作强度差异 | CellPhoneDB + ggplot2 |
| Supplementary Figure 11A–B | 热图 + 箱线图 | CS基因在28种癌种中依赖性评分(CRISPR screen)分析 | 评分越低表示越关键,比较高低组之间变化 | DepMap数据 + ggplot2 |
| Supplementary Figure 12 | 箱线图 | 比较不同癌种中CS高低组关键基因依赖性差异 | 统计检验识别关键依赖性差异 | ggplot2 |
| Supplementary Figure 13 | 箱线图 | 不同免疫刺激(TNF/IFN)对CS signature影响 | 观察不同干预对senescence signature的调节趋势 | ggplot2 |
| Supplementary Figure 14A–B | 散点图(相关分析) | senescence signature与GDSC1/2药物敏感性(IC50)的相关性 | 显著负相关表示signature越高越敏感 | Spearman + ggplot2 |
| Supplementary Figure 15A–B | 条形图 | CS高低组中MAPK通路和BCL-2家族基因表达差异 | 识别显著差异表达的抗凋亡/促凋亡基因 | limma + ggplot2 |
| Supplementary Figure 16 | 富集气泡图 | CS高组上调miRNA靶基因的KEGG通路富集分析 | 路径富集功能注释 | clusterProfiler + ggplot2 |
| Supplementary Figure 17A–B | 柱状图 | MAPK通路蛋白BRAF和ERK2在CS组间的表达水平验证 | 高CS组MAPK蛋白表达更低 | ggplot2 |
| Supplementary Figure 18 | 图形摘要(Graphical Abstract) | 总结全文研究逻辑、关键发现和应用价值 | 整合性展示研究模型与数据来源 | Figdraw 或 Illustrator |
