一、R语言学习-入门
整体规划
基础学习-数据挖掘实战-文章撰写
为什么学习R语言
- 批量计算(多数据研究)
- 重复性工作
- 功能稳定完善,免费开源适用性高
相关网站与书籍
- Bioconductor
- R Bloggers(教程大全)
- 统计之都(中文社区)
- Quick R(入门学习)
- R package documentation(很多包)
- 《R for beginners》
- 《R数据科学》
- 《R graphics cookbook》
R与R studio的安装
R中安装package
CRAN资源:
- install.packages("ggplot2")
Bioconductor资源:
- BiocManager::install("limma")
Github资源:
- library(devtools)
- install_github("immunogenomics/harmony")
二、R语言基础(一)
R studio项目代码数据管理技巧
书签代码
#### 前后各四个井号 ####注释代码
# 注释代码函数
大小写需注意,括号引号均为英文字符,函数名称内部不能有额外字符也不能少字符
"Tab"自动补齐代码
R notebook
基本使用方法与md语言一样,可以方便项目代码的注释与分类运行
R语言的数据基本类型
赋值:<-,->,=
数值Numeric
是数据挖掘的基石,可以进行数学运算
数值类型查看:
#查看数据类型(整型or双整型)
typeof() 字符串Character
使用双引号或单引号(有双引号的情况下)定义,在R中,字符串可以简单理解为字符模式的单元素向量
Eg:"内容"
#字符串长度
nchar(hello)
#大小写转换
toupper(hello) #转换大写
tolower(hello) #转换小写
#字符串连接(sep,collapse,recycle0)
paste(title,hello,sep="_")
#判断是否字符串
is.character(hello)
is.numeric() #是否数值
is.logical() #是否逻辑
#将数值转换为字符串
as.character()逻辑Logical
#直接赋值
is_female <- TRUE
is_female
is_male <- FALSE
is_male
#判断赋值
age <- 20
is_adult <- age > 18
is_adult
#例子
a<-1:100
sum(a>60)
二、R语言基础(二)
R语言的基本数据结构
Vector向量
Vector:是R中最基本的数据容器,同一向量当中的所有元素必须是相同的模式,可以是数值,字符串,逻辑值等等
Eg: a=c(0,1,2,3,4,5) / b=c('l','love','R')
如上,a变量中包含的全是数值,所以a可以用来参与数据的计算,而b中全是字符串,不能参与计算,但是可以使用专门处理字符串的函数
Vector的创建
#直接创建
a=c(1,2,3,4,5,6)
#运用冒号
b=1:10 #1到10依次取值
#使用seq函数创建
c=seq(from=1,to=10,by=2) #seq函数可以用来创建等差序列,from是起始值,to是终止值,by是生成间隔为2的向量Vector的索引与取值
格式:向量1[向量2]
如图:
如图:
Vector使用逻辑判断取值(重点)

Vector常用函数
循环补齐:对两个向量使用运算符时,如果两个向量长度不等,R会自动重复较短的向量,直到他与另一个向量长度相匹配
Eg: c(1,2,3,4)+c(1,2)输出结果——2 4 4 6
即c(1,2,3,4)+c(1,2,1,2) #其他运算符同理
> a=c(1,2,3,4,5)
#mean()函数/median() 计算向量平均值/中位值
> mean(c(1,2,3,4,5,6))
[1] 3.5
> median(c(0,1,1,2,5))
[1] 1
#sum()函数,计算向量总和
> a=c(1,2,3,4,5)
> a
[1] 1 2 3 4 5
> sum(a)
[1] 15
#which()函数,找到满足条件元素的位置
> which(a>2)
[1] 3 4 5
#sort()函数,对向量排序,默认从大到小
> sort(c(2,8,7,5,9))
[1] 2 5 7 8 9
> sort(c(2,8,7,5,9)decreasing = T)
错误: unexpected symbol在"sort(c(2,8,7,5,9)decreasing"里 #需要逗号!!!!
> sort(c(2,8,7,5,9),decreasing = T)
[1] 9 8 7 5 2
#rank()函数,对向量排序并获得排序后的序号
> b=c(2,8,7,5,9)
> b
[1] 2 8 7 5 9
> rank(b)
[1] 1 4 3 2 5
#grep()函数,抓取你想在元素中获得的对象,返回位置
> b
[1] 2 8 7 5 9
> grep(7,b)
[1] 3
#length()函数,获得向量长度
> length(b)
[1] 5
#subset()函数,对向量进行筛选
> subset(b,b>5)
[1] 8 7 9
#rep()函数,重复向量中的元素
> rep(a,2)
[1] 1 2 3 4 5 1 2 3 4 5
> rep(a,each=3)
[1] 1 1 1 2 2 2 3 3 3 4 4 4 5 5 5NA与NULL值
NA:表示缺失值
NULL:表示不存在的值,即空白
差别如下
> d=c(1,5,NA,8,12)
> mean(d)
[1] NA
> mean(d,na.rm = T)
[1] 6.5
> e=c(1,5,NULL,8,12)
> mean(e)
[1] 6.5Factor因子
因子来源于分类变量,本质上并不是数字,而是对应的分类
> b
[1] 2 8 7 5 9
> bff=factor(b)
> bff
[1] 2 8 7 5 9
Levels: 2 5 7 8 9
> str(bff)
Factor w/ 5 levels "2","5","7","8",..: 1 4 3 2 5
#这样子连续型变量就变成了分类变量Factor因子的创建
> x=c("good","bad","middle","good")
> A=factor(x)
> A
[1] good bad middle good
Levels: bad good middle
> as.numeric(A)
[1] 2 1 3 2
> B=factor(x,levels = c('good','middle','bad'))
> B
[1] good bad middle good
Levels: good middle bad
> as.numeric(B)
[1] 1 3 2 1
#factor函数可以将向量转化为因子,如果不用参数levels修改顺序的话,默认是按照首字母先后顺序排列,可以用levels参数+ordered修改顺序如何将因子转化为数值?
思路:先将因子转化为字符串,再由字符串转化为数值
> p=c(1.02,2.66,3.57,1.56,2.05)
> pp=factor(p)
> pp
[1] 1.02 2.66 3.57 1.56 2.05
Levels: 1.02 1.56 2.05 2.66 3.57
> as.numeric(pp)
[1] 1 4 5 2 3
> as.numeric(as.character(pp))
[1] 1.02 2.66 3.57 1.56 2.05
> as.character(pp) #单独转换字符串操作
[1] "1.02" "2.66" "3.57" "1.56" "2.05"Factor函数
#统计不同因子数量(同样适用于普通向量)
> xff
[1] 2 8 7 5 9
Levels: 2 5 7 8 9
> summary(xff)
2 5 7 8 9
1 1 1 1 1
> table(xff)
xff
2 5 7 8 9
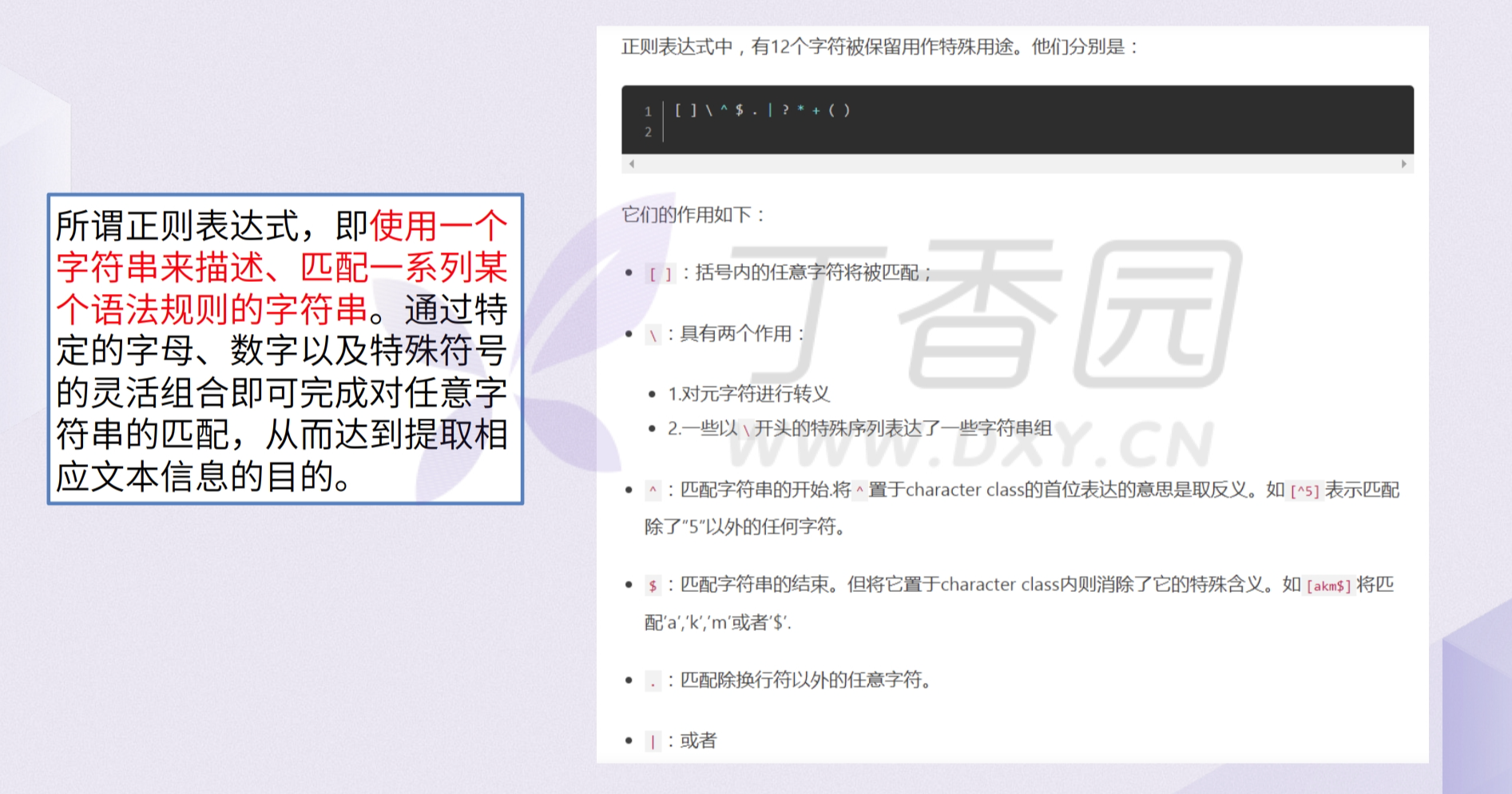
1 1 1 1 1 正则表达式应用
#练习:
> test_gene = paste0("gene",1:40)
> test_gene
[1] "gene1" "gene2" "gene3" "gene4" "gene5"
[6] "gene6" "gene7" "gene8" "gene9" "gene10"
[11] "gene11" "gene12" "gene13" "gene14" "gene15"
[16] "gene16" "gene17" "gene18" "gene19" "gene20"
[21] "gene21" "gene22" "gene23" "gene24" "gene25"
[26] "gene26" "gene27" "gene28" "gene29" "gene30"
[31] "gene31" "gene32" "gene33" "gene34" "gene35"
[36] "gene36" "gene37" "gene38" "gene39" "gene40"
#1.提取包含3的基因位置,基因
> grep(3,test_gene)
[1] 3 13 23 30 31 32 33 34 35 36 37 38 39
> grepl(3,test_gene)
[1] FALSE FALSE TRUE FALSE FALSE FALSE FALSE
[8] FALSE FALSE FALSE FALSE FALSE TRUE FALSE
[15] FALSE FALSE FALSE FALSE FALSE FALSE FALSE
[22] FALSE TRUE FALSE FALSE FALSE FALSE FALSE
[29] FALSE TRUE TRUE TRUE TRUE TRUE TRUE
[36] TRUE TRUE TRUE TRUE FALSE
> test_gene[grep(3,test_gene)]
[1] "gene3" "gene13" "gene23" "gene30" "gene31"
[6] "gene32" "gene33" "gene34" "gene35" "gene36"
[11] "gene37" "gene38" "gene39"
> test_gene[grepl(3,test_gene)]
[1] "gene3" "gene13" "gene23" "gene30" "gene31"
[6] "gene32" "gene33" "gene34" "gene35" "gene36"
[11] "gene37" "gene38" "gene39"
#2.提取不包含3的基因名字
> test_gene[-grep(3,test_gene)]
[1] "gene1" "gene2" "gene4" "gene5" "gene6"
[6] "gene7" "gene8" "gene9" "gene10" "gene11"
[11] "gene12" "gene14" "gene15" "gene16" "gene17"
[16] "gene18" "gene19" "gene20" "gene21" "gene22"
[21] "gene24" "gene25" "gene26" "gene27" "gene28"
[26] "gene29" "gene40"
> test_gene[!grep(3,test_gene)]
character(0)
> test_gene[!grepl(3,test_gene)]
[1] "gene1" "gene2" "gene4" "gene5" "gene6"
[6] "gene7" "gene8" "gene9" "gene10" "gene11"
[11] "gene12" "gene14" "gene15" "gene16" "gene17"
[16] "gene18" "gene19" "gene20" "gene21" "gene22"
[21] "gene24" "gene25" "gene26" "gene27" "gene28"
[26] "gene29" "gene40"
> !grepl(3,test_gene)
[1] TRUE TRUE FALSE TRUE TRUE TRUE TRUE
[8] TRUE TRUE TRUE TRUE TRUE FALSE TRUE
[15] TRUE TRUE TRUE TRUE TRUE TRUE TRUE
[22] TRUE FALSE TRUE TRUE TRUE TRUE TRUE
[29] TRUE FALSE FALSE FALSE FALSE FALSE FALSE
[36] FALSE FALSE FALSE FALSE TRUE
> grep(3,test_gene,value = T,invert = T)
[1] "gene1" "gene2" "gene4" "gene5" "gene6"
[6] "gene7" "gene8" "gene9" "gene10" "gene11"
[11] "gene12" "gene14" "gene15" "gene16" "gene17"
[16] "gene18" "gene19" "gene20" "gene21" "gene22"
[21] "gene24" "gene25" "gene26" "gene27" "gene28"
[26] "gene29" "gene40"
#3.提取末尾不是3的基因名字(正则表达式)
> grep("3$",test_gene,value = T,invert = T)
[1] "gene1" "gene2" "gene4" "gene5" "gene6"
[6] "gene7" "gene8" "gene9" "gene10" "gene11"
[11] "gene12" "gene14" "gene15" "gene16" "gene17"
[16] "gene18" "gene19" "gene20" "gene21" "gene22"
[21] "gene24" "gene25" "gene26" "gene27" "gene28"
[26] "gene29" "gene30" "gene31" "gene32" "gene34"
[31] "gene35" "gene36" "gene37" "gene38" "gene39"
[36] "gene40"
#4.提取末尾是3但不是gene3的基因
> grep("[0-9]3$",test_gene,value = T)
[1] "gene13" "gene23" "gene33"
> setdiff(grep("3$", test_gene,value = T),"gene3")
[1] "gene13" "gene23" "gene33"矩阵
矩阵的创建与生成
# 1.使用matrix函数
norm_data <- rnorm(15)
> norm_data
[1] 0.3130332 -0.5302314 1.6474904
[4] 0.1538946 -2.3499622 -0.3853824
[7] -1.3783288 1.5195174 -1.4208483
[10] 0.3822556 0.9944415 1.0162382
[13] 3.2295573 -1.7166756 -0.3205121
> matrix(norm_data,nrow = 5)#默认按列生成矩阵
[,1] [,2] [,3]
[1,] 0.3130332 -0.3853824 0.9944415
[2,] -0.5302314 -1.3783288 1.0162382
[3,] 1.6474904 1.5195174 3.2295573
[4,] 0.1538946 -1.4208483 -1.7166756
[5,] -2.3499622 0.3822556 -0.3205121
> matrix(norm_data,nrow = 5,byrow = T)#按行生成矩阵
[,1] [,2] [,3]
[1,] 0.3130332 -0.5302314 1.6474904
[2,] 0.1538946 -2.3499622 -0.3853824
[3,] -1.3783288 1.5195174 -1.4208483
[4,] 0.3822556 0.9944415 1.0162382
[5,] 3.2295573 -1.7166756 -0.3205121
# 2.使用rbind或者cbind合并而成
col1 <- c(1,3,8,9)
col2 <- c(2,18,27,10)
col3 <- c(8,37,267,19)
> my_matrix <- rbind(col1, col2, col3)#按行生成
> my_matrix
[,1] [,2] [,3] [,4]
col1 1 3 8 9
col2 2 18 27 10
col3 8 37 267 19
> my_matrix <- cbind(col1, col2, col3)#按列生成
> my_matrix
col1 col2 col3
[1,] 1 2 8
[2,] 3 18 37
[3,] 8 27 267
[4,] 9 10 19如何查看矩阵的信息
#获取矩阵的维度
> my_matrix
col1 col2 col3
[1,] 1 2 8
[2,] 3 18 37
[3,] 8 27 267
[4,] 9 10 19
> dim(my_matrix)
[1] 4 3
#获取矩阵的行数
> nrow(my_matrix)
[1] 4
#获取矩阵的列数
> ncol(my_matrix)
[1] 3
#矩阵转置,行列转换(矩阵转置)
> t(my_matrix)
[,1] [,2] [,3] [,4]
col1 1 3 8 9
col2 2 18 27 10
col3 8 37 267 19
#提取行名或者列名
> rownames(my_matrix)
NULL
> colnames(my_matrix)
[1] "col1" "col2" "col3"
> dimnames(my_matrix)
[[1]]
NULL
[[2]]
[1] "col1" "col2" "col3"
#修改行名(不能重复)
> rownames(my_matrix) <- c("row1", "row2", "row3", "row4")
> rownames(my_matrix) <- c("row5", "row2", "row3", "row4")
> my_matrix
col1 col2 col3
row5 1 2 8
row2 3 18 37
row3 8 27 267
row4 9 10 19
#提取矩阵信息
> my_matrix
col1 col2 col3
row5 1 2 8
row2 3 18 37
row3 8 27 267
row4 9 10 19
> my_matrix[1,3]
[1] 8
> my_matrix["row2", "col3"]
[1] 37
> my_matrix[1,]
col1 col2 col3
1 2 8
> my_matrix[,3]
row5 row2 row3 row4
8 37 267 19
#基于逻辑判断提取信息
> my_matrix[col3 > 20,]
col1 col2 col3
row2 3 18 37
row3 8 27 267
> my_matrix[col3 > 20 & col2 > 20,]
col1 col2 col3
8 27 267 数值型矩阵的常用计算
#矩阵相关计算
> my_matrix * 3
col1 col2 col3
row5 3 6 24
row2 9 54 111
row3 24 81 801
row4 27 30 57
> log10(my_matrix)
col1 col2 col3
row5 0.0000000 0.301030 0.903090
row2 0.4771213 1.255273 1.568202
row3 0.9030900 1.431364 2.426511
row4 0.9542425 1.000000 1.278754
#行列的总和
> rowSums(my_matrix)
row5 row2 row3 row4
11 58 302 38
> colSums(my_matrix)
col1 col2 col3
21 57 331
重要的矩阵处理函数apply(最基础最重要)
#apply函数主要由三部分构成:数据,行列指向,计算函数
#1代表行,2代表列
> my_matrix
col1 col2 col3
row5 1 2 8
row2 3 18 37
row3 8 27 267
row4 9 10 19
> apply(my_matrix, 1, mean)
row5 row2 row3
3.666667 19.333333 100.666667
row4
12.666667
> apply(my_matrix, 2, median)
col1 col2 col3
5.5 14.0 28.0
> apply(my_matrix, 2, sum)
col1 col2 col3
21 57 331
> apply(my_matrix, 1, function(x)sum(x))
row5 row2 row3 row4
11 58 302 38
> apply(my_matrix, 1, function(x){x[1] * 2 + x[2] * 3 + x[3] * 4})
row5 row2 row3 row4
40 208 1165 124 #练习
#提取表达量在至少一半样本中超过100且平均表达量超过200的基因
data_test <-read.table("./ReadData/raw_counts.txt",header = T,
stringsAsFactors = F,sep = "\t")
# 解法1
sel1 <- apply(data_test, 1, function(x)sum(x > 100)) >= ncol(data_test)/2
sel2 <- apply(data_test, 1, mean) > 200
data_select <- data_test[sel1 & sel2,]
# 解法2(有可能有错,因为样本数可能会被漏判)
data_select2 <- data_test[apply(data_test, 1, median) > 100 &
apply(data_test, 1, mean) > 200,]数据框(表格)
宽表更适合用于数据挖掘以及数据统计层面的分析
特点:每一行通常代表一个样本或者患者,每一列是一类数值(可能是数字也可能是逻辑也可能是字符串等),每一列都是对患者不同角度的描述;
优势:适用于日常最常见的各类因素的关联以及统计分析,比如分析不同期类型的年龄差异,不同性别的生存分析等,都是列与列之间的相关性统计;
灵活:对于日常分析中经常会增加对于患者的描述维度,只需要在表后不断增加列即可。

长表更适合用于ggplot2系列的可视化
数据框的创建
#定义基本的输入数据
> friend_names <- c("Mina", "Ella", "Anna", "Cora")
> friend_ages <- c(Mina=21, Ella=27, Anna=26, Cora=32)
> has_child <- c("TRUE", "TRUE", "FALSE", "TRUE")
# 通过data.frame创建第一个数据框
> friends <- data.frame(name=friend_names,
+ age=friend_ages,
+ child=has_child)
# 通过read.table等函数直接读入
> crc_data <- read.table("./ReadData/CRC_Metadata.txt",header = T, stringsAsFactors = F,sep = "\t")
# 从矩阵转换
> class(my_matrix)
[1] "matrix" "array"
> my_data <- as.data.frame(my_matrix)
> class(my_data)
[1] "data.frame"数据框的常用函数
# 行列名,列表,维度,行数,列数,apply均适用
> rownames(crc_data)[1:10]
[1] "1" "2" "3" "4" "5" "6" "7"
[8] "8" "9" "10"
> colnames(crc_data)
[1] "Sample_BarcodeID"
[2] "CellName"
[3] "Sample"
[4] "Tissue"
[5] "Platform"
[6] "raw.nUMI"
[7] "raw.nGene"
[8] "filter.nUMI"
[9] "filter.nGene"
[10] "ribo.per"
[11] "raw.nCD"
[12] "raw.nTF"
[13] "raw.nCyt"
[14] "raw.nLR"
[15] "Global_Cluster"
[16] "Sub_Cluster"
[17] "Sub_ClusterID"
[18] "Global_tSNE_1"
[19] "Global_tSNE_2"
[20] "Sub_tSNE_1"
[21] "Sub_tSNE_2"
[22] "integrated.Global_tSNE_1"
[23] "integrated.Global_tSNE_2"
# 大量数据查看前后几行
> head(crc_data)
> tail(crc_data)
> head(crc_data, n = 10)
> summary(crc_data) #数据总体概览(列不要太多的情况下):数字给出quantile结果,因子给出分类信息,字符串没有什么有价值的信息
数据框提取信息
# 基于行列位置,基于行列名字,基于逻辑判断均适用
# 相比矩阵matrix多了一个通过$提取某一列信息的功能
> friends$name
[1] "Mina" "Ella" "Anna" "Cora"
> friends$age
[1] 21 27 26 32
> friends[friends$age > 26,]
#使用table计数
table(crc_data$Tissue)
数据框数据改变
# 给数据框增加列
friends$married <- c("YES", "YES", "NO", "YES")dplyr数据基本处理
| 操作 | 作用 |
|---|---|
| 管道%>% | 可以一行长代码实现多步操作,省略中间数据赋值存储,大大提高效率节省空间 |
| select | 筛选列 |
| filter | 筛选行 |
| mutate | 增加或者修改列的信息 |
| rename | 重命名列 |
| arrange | 升序 |
| desc | 按照降序 |
举例说明:计算Tumor样本中每一类细胞的基因平均深度
- 提取Tumor样本中的CD8 T cell
- 并计算每个细胞中每个基因的均测到的UMI
- 只保留样本名,细胞名,细胞类型,UMI,gene数量,平均数量
- 并将名字都改为首字母大写无小数点
- 最后按照深度从高到低排序
> table(crc_data$Global_Cluster)
B cell CD4 T cell CD8 T cell
9791 11028 8527
ILC Myeloid cell
1846 12625
> crc_data %>%
+ filter(Tissue == "T", grepl("CD8",Global_Cluster)) %>%
+ mutate(UMIratio = round(filter.nUMI/filter.nGene,3)) %>%
+ select(Sample,CellName,Global_Cluster,
+ filter.nUMI,filter.nGene,UMIratio) %>%
+ rename(Ngene = filter.nGene,
+ Numi = filter.nUMI,
+ CellType = Global_Cluster) %>%
+ arrange(desc(UMIratio)) -> crc_filtered
> View(crc_filtered)列表
一些数据结构的基本操作
# 创建
> ele_1 <- "Sophia"
> ele_2 <- c("Mina","Ella","Darren","Rita")
> ele_3 <- matrix(rnorm(20),nrow = 5)
> ele_4 <- data.frame(Gene = c("EGFR","KRAS","SMAD4"),
+ Mutation = c("WT","Mut","WT"),
+ Patient = paste0("Pat",1:3))
> test_list <- list(ele_1,ele_2,ele_3,ele_4)
> test_list <- list(Doctor = ele_1,
+ Friend = ele_2, Info = ele_3,
+ Patient = ele_4)
> print(test_list) #适用于所有数据结构的输出函数(但是一般不要这么输出)
$Doctor
$Doctor
[1] "Sophia"
$Friend
[1] "Mina" "Ella" "Darren" "Rita"
$Info
[,1] [,2] [,3]
[1,] 0.6722184 1.74080872 0.6808691
[2,] -1.1428504 -0.01948443 -2.6503907
[3,] -0.7605064 2.57900651 -0.3582360
[4,] -1.5104113 0.39095948 -1.2302126
[5,] 0.8092676 -0.25098669 0.5521289
[,4]
[1,] 0.1547392
[2,] -0.7443876
[3,] -0.5636841
[4,] 0.6671895
[5,] -0.7839512
$Patient
Gene Mutation Patient
1 EGFR WT Pat1
2 KRAS Mut Pat2
3 SMAD4 WT Pat3
#命名
> names(test_list)
[1] "Doctor" "Friend" "Info" "Patient"
# 更新
> names(test_list)
[1] "Doctor" "Friend" "Info" "Patient"
> names(test_list)[1] <- "Mother"
> test_list[1]
$Mother
[1] "Sophia"
> test_list[[1]]
[1] "Sophia"
> class(test_list[1])
[1] "list"
> class(test_list[[1]])
[1] "character"
> test_list[[2]][1]
[1] "Mina"
> test_list[[2]][1] <- "Anna"
# 合并
> new_list <- list(Age = c(21,37,15))
> merged_list <- c(test_list,new_list)
> print(merged_list)
$Mother
[1] "Sophia"
$Friend
[1] "Anna" "Ella" "Darren" "Rita"
$Info
[,1] [,2] [,3]
[1,] 0.6722184 1.74080872 0.6808691
[2,] -1.1428504 -0.01948443 -2.6503907
[3,] -0.7605064 2.57900651 -0.3582360
[4,] -1.5104113 0.39095948 -1.2302126
[5,] 0.8092676 -0.25098669 0.5521289
[,4]
[1,] 0.1547392
[2,] -0.7443876
[3,] -0.5636841
[4,] 0.6671895
[5,] -0.7839512
$Patient
Gene Mutation Patient
1 EGFR WT Pat1
2 KRAS Mut Pat2
3 SMAD4 WT Pat3
$Age
[1] 21 37 15
# 转换为vector(选择合适的机会使用)
> View(unlist(test_list))
> length(test_list)
[1] 4
> print(test_list)
$Mother
[1] "Sophia"
$Friend
[1] "Anna" "Ella" "Darren" "Rita"
$Info
[,1] [,2] [,3]
[1,] 0.6722184 1.74080872 0.6808691
[2,] -1.1428504 -0.01948443 -2.6503907
[3,] -0.7605064 2.57900651 -0.3582360
[4,] -1.5104113 0.39095948 -1.2302126
[5,] 0.8092676 -0.25098669 0.5521289
[,4]
[1,] 0.1547392
[2,] -0.7443876
[3,] -0.5636841
[4,] 0.6671895
[5,] -0.7839512
$Patient
Gene Mutation Patient
1 EGFR WT Pat1
2 KRAS Mut Pat2
3 SMAD4 WT Pat3R语言数据读写与存取
#指定数据路径
filename <- "./ReadData/GSE107754_series_matrix.txt"
data_series <- read.table(filename,
comment.char = "!",
header = T,
nrows = 10,
row.names = 1)
data_series <- read.table(filename,
header = T,
nrows = 10,
row.names = 1,
skip = 75)
#读取csv文件
data_csv <- read.csv("./ReadData/GSE138042_mRNA_seq_thyroid.csv",row.names = 1)
View(data_csv[1:10,])
#读取excel文件
data_xlsx <- openxlsx::read.xlsx("./ReadData/Excel数据练习.xlsx",sheet = 2,rowNames = T,rows = 1:10)
##写入
#写到txt中
write.table(data_series,file = "./ReadData/Write_table.txt",
sep = "\t",row.names = T,col.names = T,
quote = T)
#写到csv中
write.csv(data_series,file = "./ReadData/Write_csv.csv",
row.names = F)
#写到excel中
write.xlsx(data_series,file = "./ReadData/Write_excel.xlsx",
sheetName = "test")
##save 与 load
#Save可以保存一个或者多个数据到同一个文件中
save(data_series,data_xlsx,file = "./ReadData/Saved_data.RData")
#通过load函数即可快速加载RData中的数据
load("./ReadData/Saved_data.RData")
#saveRDS 与 readRDS可以存取一个R的对象,且可重新命名
saveRDS(data_series,file = "./ReadData/Saved_rds.rds")
data_rds <- readRDS("ReadData/Saved_rds.rds")
数据结构之间的转换、高频函数(总结补充)
数据结构的判断与转换:is、as
- 数据类型判断:class(够用)
- 特定类型判断:is.na(高频)、is.data.frame等
- 数据类型转换:as.data.frame、as.character等
# 理解is.na的使用场景
> sum(is.na(tcga_clic_ana$Bilirubin))
[1] 83
# 计算Bilirubin中NA的占比
> sum(is.na(tcga_clic_ana$Bilirubin))/nrow(tcga_clic_ana)
[1] 0.1769723
# 在数据中存在NA时该如何计算?
> mean(tcga_clic_ana$Bilirubin,na.rm = T) #na.rm可以去除NA
# 最常用的矩阵转数据框
> exam_matrix <- matrix(rnorm(20),nrow = 4)
> class(exam_matrix)
[1] "matrix" "array"
> exam_df <- as.data.frame(exam_matrix)
> class(exam_df)
[1] "data.frame"
最容易出错的factor转数字
> a <- factor(round(rnorm(5),2))
> a
[1] -0.07 0.15 0.74 0.75 0
Levels: -0.07 0 0.15 0.74 0.75
> mean(a)
[1] NA
##Warning message:
##In mean.default(a) : 参数不是数值也不是逻辑值:回覆NA
> as.numeric(a) #这是不正确的
[1] 1 3 4 5 2
> as.numeric(as.character(a)) #这个才是正确的
[1] -0.07 0.15 0.74 0.75 0.00
> as.numeric(as.vector(a)) #这个也是正确的
[1] -0.07 0.15 0.74 0.75 0.00
数据挖掘高频使用的函数总结(表格)
| 常用函数 | 作用 |
|---|---|
| 已学函数--基本 | |
| length() | 计算向量的长度 |
| nchar() | 计算字符串的字符数 |
| toupper() | 字母的大写转换 |
| tolower() | 字母的小写转换 |
| dim() | 查看矩阵或者数据框的行列维度 |
| nrow() | 查看矩阵或者数据框的行数 |
| ncol() | 查看矩阵或者数据框的列数 |
| names() | 数据的name,colnames,rownames等 |
| head() | 查看数据的前几行,同样的tail后几行 |
| 已学函数--常用 | |
| table() | 分组统计每一类的数量 |
| summary() | 整体查看数据的特征与分布 |
| read.table() | 数据读写函数必备,save/load,read.xlsx等 |
| data.table::fread() | |
| 较难函数--高频使用 | |
| apply() | 按照行列进行计算 |
| aggregate() | 分组进行统计 |
| merge() | 多数据框合并 |
| dplyr::select() | dplyr包中的几个必备函数select,filter,mutate,rename,left_join,arrange,%>%等 |
补充
特定包内函数的使用
#特定包内函数的使用
data.table::fread() #包::函数(),可以调用某一个包里面的函数
BiocManager::install()
read.xlsx() # library(openxlsx),先加载包再调用函数序列的交集、差集、并集
a <- 1:10
b <- 5:14
# 交集
intersect(a,b)
# 差集
setdiff(a,b) #a在前,a里面有b里面没有的
setdiff(b,a) #b在前,b里面有a里面没有的
# 并集
union(a,b)
# 去除重复
unique(c(a,b))多数据框的合并merge/join(重难点)
通过merge函数可将两个数据框按照某一列或者某几列进行合并添加列;
join函数(来自dplyr包)共有四个姐妹函数,功能同merge;
merge函数
> df_1 <- data.frame(
+ ID = c(1:4,6),
+ Name = c("Jim","Tony","Lisa","Rita","Tom"),
+ Gender = c("M","M","F","F","M"))
> df_2 <- data.frame(
+ IDnum = 1:5,
+ Age = c(22,15,19,59,10))得出的数据框如下


合并操作
# 合并操作1:依照ID匹配合并,不匹配的去除
> merge(df_1, df_2, by = "ID")
# 合并操作2:依照ID匹配合并,保留x(即df_1的数据)
> merge(df_1, df_2, by = "ID",all.x = T)
# 合并操作3:依照ID匹配合并,保留y(即df_2的数据)
> merge(df_1, df_2, by = "ID",all.y = T)
# 合并操作4:依照ID匹配合并,保留所有数据
> merge(df_1, df_2, by = "ID",all = T)
#注意:若匹配列名不一样的时候,可以指定列名匹配
> merge(df_1,df_2,by.x = "ID",by.y = "IDnum")操作4得出数据框如下

join函数(dplyr)
# 等价的join函数
> inner_join(df_1,df_2,by = "ID") #同上操作1
> left_join(df_1,df_2,by = "ID") #同上操作2
> right_join(df_1,df_2,by = "ID") #同上操作3
> full_join(df_1,df_2,by = "ID") #同上操作4aggregate函数
# 计算不同性别患者的平均年龄
aggregate(Age ~ Gender,data = df_merge,mean)翻译:按照gender对age进行分组,然后计算age里面每一组的平均值
三、R函数学习1
R语言中的循环函数——for/while
for循环
for (var in seq) expr
#var 表示循环变量
#seq 表示向量
#expr 为执行的语句
# 依照seq的顺序依次执行expr
#例子:计算1~100相加的和
> i.sum <- 0
> for(i in 1:100){i.sum = i.sum + i}
> i.sum
[1] 5050
while循环
(谨慎:会无限循环导致电脑死机)
while(test_expression){statement}
#test_expression 为判断条件
#statement 为执行语句
# 判断结果为T啧执行一次statement语句,然后继续判断
#例子:计算1~100相加的和
> i.sum <- 0
> i = 1
> while(i <=100){i.sum = i.sum + i
+ i = i+1 }
> i.sum
[1] 5050
> i
[1] 101矩阵维度的循环示例
# 计算矩阵中每一列的中位值
> exam_matrix <- matrix(rnorm(100), ncol = 10)
> median_out <- rep(0,ncol(exam_matrix))
> for(i in 1:ncol(exam_matrix)){
+ median_out[i] <- median(exam_matrix[,1])
+ }
> median_out
[1] -0.3552402 -0.3552402 -0.3552402 -0.3552402 -0.3552402
[6] -0.3552402 -0.3552402 -0.3552402 -0.3552402 -0.3552402
# 计算矩阵中每一列的中位值和平均值
> df_out <- data.frame(Median = rep(0,ncol(exam_matrix)),Mean = 0)
> for(i in 1:ncol(exam_matrix)){
+ df_out$Median[i] <- median(exam_matrix[,i])
+ df_out$Mean[i] <- mean(exam_matrix[,i])
+ }
#注意:每次循环之前先把要存储的数据提前生成特定的结构,会大大提高循环的计算效率循环中的判断(重点)——if/else
==:比较符号,表示两者大小相等
%%:相除取余数
!=:不等于
break:跳出循环
#例子:计算1-100中偶数的和
# 写法一:理解执行中的判断
> i.sum <-0
> for (i in 1:100) {
+ if(i %% 2 == 0){
+ i.sum = i.sum + i
+ }else{
+ next
+ }
+ }
> i.sum
[1] 2550
# 写法二:理解next
> i.sum <- 0
> for(i in 1:100){
+ if(i %% 2 != 0){
+ next
+ }
+ i.sum = i.sum + i
+ }
# 写法三
> sum(seq(0,100,by = 2))
[1] 2550循环中的复杂判断if...else
#if...else循环(其中else也可以为空也可以多个,多个else则中间的需要写成else if)
#计算1-100中小于50的奇数和大于等于50的偶数之和
> i.sum <- 0
> con_count <- 0 #计算执行了多少次
> for(i in 1:100){
+ if(i < 50 & i %% 2 != 0){
+ i.sum = i.sum + i
+ }else if(i >= 50 & i %% 2 == 0){
+ i.sum = i.sum + i
+ }else{
+ next
+ }
+ con_count <- con_count + 1
+ }
> i.sum
[1] 2575
> con_count #计算执行了多少次
[1] 51
# 验证函数
> sum(seq(1,50,by = 2),seq(50,100,by = 2))
## if...else的简体ifelse,在数据挖掘中必用(二分类判断)
> i <- 101
> ifelse(i %% 2 == 0,"偶数","奇数")
[1] "奇数"
> i <- 100
> ifelse(i %% 2 == 0,"偶数","奇数")
[1] "偶数"
> i <- 1:10
> i
[1] 1 2 3 4 5 6 7 8 9 10
> table(ifelse(i >= 5,"Old","Young"))
Old Young
6 4 练习:
计算raw_count.txt文件中每一个样本中最高表达的10个基因,并保存为数据框
# 第一步:读入数据,并简单查看确认读入的数据无误
> raw_count <- read.table("./ReadData/raw_counts.txt",
+ header = T,stringsAsFactors = F)
> View(raw_count)
# 第二步:创建结果保存的数据框
> out_data <- as.data.frame(matrix("gene",nrow = 10,ncol = ncol(raw_count)))
> colnames(out_data) <- colnames(raw_count)
# 第三步:按照列名逐个遍历对应的列,分别提取最高的10个基因保存
for(sam_name in colnames(raw_count)){
# 1. 提取对应列的数据
tmp_data <- raw_count[,sam_name]
# 2. 将该列的数据按照从大到小排序
tmp_order <- order(tmp_data,decreasing = T)
# 3. 提取排序中前10个数据所在的位置
tmp_order <- tmp_order[1:10]
# 4. 按照这个位置去提取对应的基因
tmp_gene <- rownames(raw_count)[tmp_order]
# 上面四步合并一行
## tmp_gene <- rownames(raw_count)[order(raw_count[,sam_name],decreasing = T)[1:10]]
# 5. 将得到的10个基因替换存储的结果
out_data[,sam_name] <- tmp_gene
cat(sam_name," finished\n")
}
注意:
- 用不同方式读入的数据格式可能不同,所以数据分析之前一定要对自己的数据了如指掌,查看数据是必不可少的步骤;
- 如果在for循环之前明确知道最终结果是什么形式,提前生成相似形式的数据,最终再逐条替换结果;
- 建议将复杂代码分步拆解完成,学会debug。
R语言中的循环函数——apply
回顾

实例练习
# 读入CRC数据
crc_data <- read.table("./ReadData/CRC_Metadata.txt",
header = T,
stringsAsFactors = F,sep = "\t")
# 查看数据
dim(crc_data)
View(crc_data)
View(crc_data %>% select(2:7,Global_Cluster))
table(crc_data$Tissue,crc_data$Global_Cluster)
table(crc_data$Tissue)
# 问题:统计每一类Tissue中不同细胞类型的占比
# 计算每一类Tissue的总细胞数(分母)
> tissue_total<- data.frame(table(crc_data$Tissue))
> tissue_total
Var1 Freq
1 N 12657
2 P 20466
3 T 10694
> colnames(tissue_total) <- c("Tissue","Total")
> tissue_total
Tissue Total
1 N 12657
2 P 20466
3 T 10694
#计算每一类Tissue的每类细胞数量(分子)
> tissue_cellct <- data.frame(table(crc_data$Tissue, crc_data$Global_Cluster))
> colnames(tissue_cellct) <- c("Tissue","Cell","Count")
# 合并分子与分母
> tissue_cellct <- merge(tissue_cellct, tissue_total) %>% mutate(Percentage = round(Count/Total,3) * 100)
##### 用apply来完成
> tissue_cellct <- data.frame(table(crc_data$Tissue,
+ crc_data$Global_Cluster))
> tissue_cellct$Total <- apply(tissue_cellct, 1, function(x){
+ nrow(crc_data[crc_data$Tissue == x[1],])
+ })
> tissue_cellct <- tissue_cellct %>%
+ mutate(Percentage = round(Freq/Total,3) * 100)使用ggplot2画图展示对比(承接上一实例)
此处代码暂不做要求,以后会学到的
# 用ggplot2画图并将图的结果保存到变量p
p <- ggplot(tissue_cellct,aes(x = Var1, y = Percentage, fill = Var2)) + geom_bar(stat = "identity",position = "stack") +
theme_classic() + scale_fill_nejm() +
theme(axis.title.x = element_blank()) +
labs(fill = "CellType", y = "Percentage(%)") +
geom_text(aes(label = paste0(Percentage,"%")),
position = position_stack(vjust = .5))
# 将图的结果保存到pdf文件中
ggsave(filename = "./Out/CRC_tissue_cell_fraction.pdf",
plot = p,width = 4.5,height = 4)
使用R语言生成的第一幅统计图表,具有重要意义(doge)
练习解析

R语言中的自定义函数——function
function函数基本语法规则
#函数的主要结构构成
function_name <- function(arg_1, arg_2, ...){
Funtion body
}自定义函数的基本结构包括:
- 函数名称:字母(大小写均可),下划线,数字(不能在首位),英文句号均可以;
- function函数:function()
- 参数:可以一个也可以多个,参数也可以提供默认值;
- 函数主体:识别参数并进行各种计算的语句集合(可以是一条也可以是多条,数据只能来自参数)
- 函数返回结果:return()函数,或者最后一步计算结果不给赋值
#例子1
> minusMean2 <- function(x) {
+ a.mean <- mean(x)
+ result <- x - a.mean
+ return(result)
+ }
> minusMean2(1:10)
[1] -4.5 -3.5 -2.5 -1.5 -0.5 0.5 1.5 2.5 3.5
[10] 4.5
#例子2(多参数)
# 给定向量,乘以系数,减去特定值
> minusMean3 <- function(x, weight = 1.2, fee = 200){
+ x.new <- x * weight
+ x.new - fee
+ }
> salary <- c(4000,4500,4300,8000)
> minusMean3(salary)
[1] 4600 5200 4960 9400
#对参数进行修改
minusMean3(salary)
minusMean3(x = salary)
minusMean3(x = salary, weight = 1.5)
minusMean3(x = salary, fee = 300)
minusMean3(salary,1.5,300)#正确
minusMean3(salary,300,1.5)#错误- 多参数:如果函数具有多个参数,调用的时候建议给定参数名称,否则会按照提供的位置进行调用;
- 默认参数:调用时可以不提供,如果没有默认值的参数必须提供
理解内部语句连接(函数中嵌套控制判断)
# 工资计算函数中只有当工资超过5000才扣除一笔工会费
salaryCal <- function(sa, weight = 1.2, over = 5000, fee = 200){
sa.total = sa * weight
sa.minus = ifelse(sa.total > over,fee,0)
sa.rest = sa.total - sa.minus
out <- data.frame(Basis = sa,
Total = sa.total,
Minus = sa.minus,
Rest = sa.rest)
return(out)
}
sa_left <- salaryCal(salary)
sa_left <- salaryCal(salary,weight = 1.5,over = 6000,fee = 250)小练习
# 0-1标准化:x - 最小值,然后除以极差(最大值-最小值)
Normalize01 <- function(x){
if(is.numeric(x)){
x.min <- min(x, na.rm = T)
x.max <- max(x, na.rm = T)
return((x - x.min)/(x.max - x.min))
}else{
stop("Data must be numeric")
}
}
> Normalize01(letters[1:4])
Error in Normalize01(letters[1:4]) : Data must be numeric
> Normalize01(1:10)
[1] 0.0000000 0.1111111 0.2222222 0.3333333
[5] 0.4444444 0.5555556 0.6666667 0.7777778
[9] 0.8888889 1.0000000
> Normalize01(c(1:10,NA))
[1] 0.0000000 0.1111111 0.2222222 0.3333333
[5] 0.4444444 0.5555556 0.6666667 0.7777778
[9] 0.8888889 1.0000000 NA
# Z-score:x - 均值,然后除以标准差
Zscore <- function(x){
if(is.numeric(x)){
x.mean <- mean(x,na.rm = T)
x.sd <- sd(x,na.rm = T)
return((x - x.mean)/x.sd)
}else{
stop("Data must be numeric")
}
}
> Zscore(1:10)
[1] -1.4863011 -1.1560120 -0.8257228 -0.4954337
[5] -0.1651446 0.1651446 0.4954337 0.8257228
[9] 1.1560120 1.4863011
> scale(1:10) # R自带zscore函数进阶自定义函数(统计分析)【重要!!!】
- [ ] 构建并管理自己的代码库
- [ ] R语言中常用的统计分析方法
- [ ] 快速自动化完成常见统计分析
自定义代码库——数据挖掘必备
- 新建一个Codes文件夹用来存储所有的成熟code
- 新建一个R代码文件
- 将写好并测试成功的函数保存到该文件中,并写清楚函数的具体功能
# 通过source加载之后再调用
source("./Codes/Zscore.R")
Zscore(1:10)一些数据挖掘中基础的统计分析

连续变量
- 在一定区间内可以任意取值的即为连续变量
- 如:年龄,体重,基因表达量,血常规的一些检测结果等等
分类变量
- 变量值是定性的,取值为分类数据
- 如:性别,肿瘤分级,是否吸烟等
生存时间
- 由随访时间和随访状态两个数据组成,随访时间代表从对患者的追踪到最后一次随访的时间间隔(也有用随访开始和随访结束两个点来表示)
- 随访状态为最后一次随访时是否发生观察事件,例如随访患者的死亡状态,如果死亡则为1,未死亡则为0,如果失访或者因其他原因死亡则即为删失也是0;
批量进行分析统计
代码实例(body部分大致看看即可)

#data:待计算的数据(data.frame格式)
#gene_cat:分类变量1
#gene_con:连续变量1
#clinical_cat:分类变量2(可与gene_cat相同)
#clinical_con:连续变量2(可与gene_con相同)
#survival:生存类型
#clinical_order:有序的连续变量
#adjust_gene:校正的变量1(gene_con/cat子集)
#adjust_parameter:校正因素集合
clinical.matrix<-function(data,gene_cat=vector(),
gene_con=vector(),
clinical_cat=vector(),
clinical_order=vector(),
clinical_con=vector(),
survival=vector(),
adjust_gene=vector(),
adjust_parameter=vector()){
library(survival)
p=vector()
for (i in gene_cat){
for(m in clinical_con){
eval(parse(text=paste(
"data_tmp=data[!is.na(data$`",i,"`)&!is.na(data$`",m,"`),]",
sep=""
)))
flag=vector()
eval(parse(text=paste(
"flag=length(unique(data_tmp$`",i,"`))",
sep=""
)))
if(flag>2){
eval(parse(text=paste(
"p=c(p,summary(aov(`",m,"`~`",i,"`,data=data_tmp))[[1]]$`Pr(>F)`[1])",
sep=""
)))
}else if(flag==2){
eval(parse(text=paste(
"p=c(p,wilcox.test(data_tmp$`",m,"`[data_tmp$`",i,"`==unique(data_tmp$`",i,"`)[1]],data_tmp$`",m,"`[data_tmp$`",i,"`==unique(data_tmp$`",i,"`)[2]])$p.value)",
sep=""
)))
}else{
p=c(p,NA)
}
}
for(k in clinical_order){
eval(parse(text=paste(
"data_tmp=data[!is.na(data$`",i,"`)&!is.na(data$`",k,"`),]",
sep=""
)))
flag=vector()
eval(parse(text=paste(
"flag=length(unique(data_tmp$`",i,"`))",
sep=""
)))
if(flag>2){
eval(parse(text=paste(
"p=c(p,summary(aov(as.numeric(",k,")~`",i,"`,data=data_tmp))[[1]]$`Pr(>F)`[1])",
sep=""
)))
}else if(flag==2){
eval(parse(text=paste(
"p=c(p,cor.test(as.numeric(data_tmp$`",i,"`),as.numeric(data_tmp$",k,"))$p.value)",
sep=""
)))
}else{
p=c(p,NA)
}
}
for(n in clinical_cat){
if(i==n){p=c(p,NA);next}
eval(parse(text=paste(
"data_tmp=data[!is.na(data$`",i,"`)&!is.na(data$`",n,"`),]",
sep=""
)))
if(nrow(data_tmp)==0){p=c(p,NA);next}
flag=vector()
eval(parse(text=paste(
"flag=all(length(unique(data_tmp$`",i,"`))>1,length(unique(data_tmp$`",n,"`))>1)",
sep=""
)))
fisher_flag=vector()
eval(parse(text=paste(
# "fisher_flag=any(all(as.vector(acast(data_tmp,`",i,"`~`",n,"`,value.var='`",n,"`',length))>4),length(unique(data_tmp$`",n,"`))*length(unique(data_tmp$`",i,"`))>10)",
"fisher_flag=length(unique(data_tmp$`",n,"`))*length(unique(data_tmp$`",i,"`))>10",
sep=""
)))
if(flag==TRUE){
if(fisher_flag==TRUE){
eval(parse(text=paste(
"p=c(p,chisq.test(data_tmp$`",i,"`,data_tmp$`",n,"`)$p.value)",
sep=""
)))}else{
eval(parse(text=paste(
"p=c(p,fisher.test(data_tmp$`",i,"`,data_tmp$`",n,"`)$p.value)",
sep=""
)))
}
}else{
p=c(p,NA)
}
}
for (s in survival){
eval(parse(text=paste(
"data_tmp=data[!is.na(data$`",i,"`)&!is.na(data$`",s,"`)&!is.na(data$`",s,".status`),]",
sep=""
)))
flag=vector()
eval(parse(text=paste(
"flag=length(unique(data_tmp$`",i,"`))>1",
sep=""
)))
if(flag==TRUE){
adjust=(i %in% adjust_gene)
if(adjust==TRUE){
eval(parse(text=paste(
"p=c(p,summary(coxph(Surv(`",s,"`,`",s,".status`)~`",i,"`+",paste(adjust_parameter,collapse = '+'),",data=data))$coefficients[1,5])",
sep=""
)))
}else{
eval(parse(text=paste(
"p=c(p,1-pchisq(survdiff(Surv(`",s,"`,`",s,".status`)~data$`",i,"`,data=data)$chisq,length(survdiff(Surv(`",s,"`,`",s,".status`)~data$`",i,"`,data=data)$n)-1))",
sep=""
)))
}
}else{
p=c(p,NA)
}
}
}
for (i in gene_con){
for(m in c(clinical_con,clinical_order)){
if(i==m){p=c(p,NA);next}
eval(parse(text=paste(
"data_tmp=data[!is.na(data$`",i,"`)&!is.na(data$`",m,"`),]",
sep=""
)))
if(nrow(data_tmp)==0){
p=c(p,NA)
}else{
eval(parse(text=paste(
"p=c(p,cor.test(as.numeric(data_tmp$`",i,"`),as.numeric(data_tmp$`",m,"`),method='spearman')$p.value)",
sep=""
)))
}
}
for(n in clinical_cat){
eval(parse(text=paste(
"data_tmp=data[!is.na(data$`",i,"`)&!is.na(data$`",n,"`),]",
sep=""
)))
eval(parse(text=paste(
"flag=length(unique(data_tmp$`",n,"`))",
sep=""
)))
if(flag==2){
eval(parse(text=paste(
"p=c(p,wilcox.test(data_tmp$`",i,"`[data_tmp$`",n,"`==unique(data_tmp$`",n,"`)[1]],data_tmp$`",i,"`[data_tmp$`",n,"`==unique(data_tmp$`",n,"`)[2]])$p.value)",
sep=""
)))
}else if(flag>2){
text=paste(
"p=c(p,summary(aov(`",i,"`~`",n,"`,data=data_tmp))[[1]]$`Pr(>F)`[1])",
sep=""
)
eval(parse(text=text))
}else{
p=c(p,NA)
}
}
for (s in survival){
eval(parse(text=paste(
"data_tmp=data[!is.na(data$`",i,"`)&!is.na(data$`",s,"`)&!is.na(data$`",s,".status`),]",
sep=""
)))
flag=vector()
eval(parse(text=paste(
"flag=length(unique(data_tmp$`",i,"`))>1",
sep=""
)))
if(flag==TRUE){
if(i %in% adjust_gene){
eval(parse(text=paste(
"p=c(p,summary(coxph(Surv(`",s,"`,`",s,".status`)~`",i,"`+",paste(adjust_parameter,collapse = '+'),",data=data))$coefficients[1,5])",
sep=""
)))
}else{
eval(parse(text=paste(
"p=c(p,summary(coxph(Surv(`",s,"`,`",s,".status`)~`",i,"`,data=data))$coefficients[1,5])",
sep=""
)))
}
}else{
p=c(p,NA)
}
}
}
p.data=data.frame(clinical=rep(c(clinical_con,clinical_order,clinical_cat,survival),length(c(gene_cat,gene_con))),
gene=rep(c(gene_cat,gene_con),each=length(c(clinical_con,clinical_order,clinical_cat,survival))),p=p)
p.data$clinical=factor(p.data$clinical,levels=c(clinical_con,clinical_order,clinical_cat,survival))
p.data$gene=factor(p.data$gene,levels=c(gene_cat,gene_con))
p.data$p.log=-log(p.data$p,base=10)
p.data$p.log[p.data$p.log>4]=4
return(p.data)
}
使用此代码对数据进行批量处理:
# 加载分析函数
source("./Codes/clinical_matrix3.R")
# 读入测试数据
tcga_test <- read.xlsx("./ReadData/TCGA_clinical_test.xlsx",sheet = 1)
# 查看并研究测试数据以便进行调整
View(tcga_test)
# 对数据进行一些修饰之后再做计算
tcga_test <- tcga_test %>%
rename(OS.status = OS) %>%
rename(OS = OS.time)
# 把一些空值变成NA,以免空值自成一类参与计算
tcga_test[tcga_test == ""] <- NA
# 检查一些要计算的分类变量是否合理,比如T分期中T1a和T1b可以合并成T1
table(tcga_test$Gender)
table(tcga_test$Inflamation)
table(tcga_test$Tstage)
quantile(tcga_test$Bilirubin,na.rm = T)
tcga_test <- tcga_test %>% mutate(Tstage = case_when(
grepl("T1",Tstage) ~ "T1",
grepl("T2",Tstage) ~ "T2",
grepl("T3",Tstage) ~ "T3",
grepl("T4",Tstage) ~ "T4",
T ~ NA_character_
))
gene_cat <- c("Gender","Grade","Mstage","Nstage","Tstage","Vascular",
"Inflamation")
gene_con <- c("Age","Bilirubin")
cli_cat <- gene_cat
cli_con <- gene_con
sur_type <- c("OS")
cli_test <- clinical.matrix(data = tcga_test,gene_cat = gene_cat,
gene_con = gene_con,
clinical_cat = cli_cat,
clinical_con = cli_con,
survival = sur_type)
View(cli_test)批量进行可视化分析(我大受震撼)
#plot.data.past <- plot.data
plot.data <- cli_test[!is.na(cli_test$p),]
plot.data$p <- round(plot.data$p,2)
for(i in 1:nrow(plot.data)){
if(plot.data[i,"p.log"] >= 1.3 & plot.data[i,"p.log"]<3){
plot.data$p.log[i] = 3
}
}
plot.data$p[plot.data$p > 0.1] <- NA
plot.data <- plot.data %>%
mutate(clinical = factor(clinical,
levels = c(gene_con,gene_cat,sur_type),
ordered = T),
gene = factor(gene,levels = c(gene_con,gene_cat),ordered = T))
p <- ggplot(data=plot.data,aes(x=clinical,y=gene,label=p))+
geom_tile(aes(fill=p.log),colour="white")+
scale_fill_gradient2(low="darkgreen",mid="yellow",midpoint=2,high="red")+
geom_text(size = 2.5) +
theme(axis.text.x=element_text(size=10,angle = 45,vjust = 1,hjust = 1),
axis.text.y=element_text(size=10),
axis.title=element_blank(),
axis.ticks= element_blank(),
text = element_text(size=10)) +
labs(fill = "-lop(P)")
ggsave(filename = "./Out/Clinical_correlation.pdf",plot = p)得出图片

这便是数据挖掘的普筛
- 图中格子里面的数字代表p值,越接近红色则说明p值越显著,低于0.2的在图中均进行了展示;
- 每一个格子的p值代表横轴与纵轴两个因素的统计检验结果,例如左上角的0.04代表Inflamation与Age是有显著相关性的;
- 最后一列代表预后分析,说明Age,Mstage,Nstage,Tstage均与OS有显著相关性,其他因素未见显著影响
如何用R获取公共数据,管理数据
用R语言自动下载数据——download.file函数
?download.file
rna_url <- "https://gdc.xenahubs.net/download/TCGA-READ.htseq_fpkm-uq.tsv.gz"
# 这里destfile一定要保持与原始数据的格式一致(文件名后缀)
download.file(rna_url,destfile = "./ReadData/tmp.tcv.gz")- url:要下载的数据链接
- destfile:下载之后存储的文件名以及路径
用R语言自动完成大部分重复性工作
- 方法一:把明确重复性的过程打包成函数,写成function
- 方法二:把不完全重复性的工作写成相对模块化的多段代码
实例:(该函数可自动下载RNA表达谱以及临床特征数据并进行简单的清洗与保存)
xena数据库:几乎包含了所有TCGA的level3数据,且格式统一内容完整,针对不同癌种相同分析时非常适用
# 该函数可自动下载xena中的RNA表达谱以及临床特征数据,并对临床数据可以做简单的清洗,例如将常用列改名,不常用列删除等,并且会将下载清洗之后的数据之间保存为Rdata,其中RNA表达量数据则会保存为矩阵
Downld_xena <- function(url1 = NULL, outname = "download.txt", destdir = "./", datatype = "rna", clean = T,RMraw = T){
# 检查两个包的可用性dplyr以及tibble
if(!require(dplyr)){
install.packages("dplyr")
}
if(!require(tibble)){
install.packages("tibble")
}
# 目前只支持最常用的rna以及clinical数据下载
if(!datatype %in% c("rna","clinical","survival")){
stopifnot("datatype can only be one of rna or clinical")
}
# 提示URL必须是非空字符串
if(is.null(url1) | !is.character(url1)){
stopifnot("You should check the url1 parameter")
}
# 下载数据到特定目录中
tmp_file <- paste(destdir,rev(strsplit(rna_url,"/")[[1]])[1],sep = "/")
download.file(url1,destfile = tmp_file)
# cat("Finished download file")
tmp_data <- data.table::fread(tmp_file)
# 如果是rna则只是保存成Rdata,如果是clinical则进行简单的清洗
if(datatype == "clinical" & clean){
tmp_data <- tmp_data %>%
dplyr::rename(Age = age_at_initial_pathologic_diagnosis,
Gender = gender.demographic,
PID = submitter_id,
Sample_ID = submitter_id.samples,
Sample_Type = sample_type.samples,
Stage = tumor_stage.diagnoses) %>%
dplyr::select(-c(starts_with("additional"),starts_with("year"),
starts_with("bcr"),starts_with("pathology"),
starts_with("days"),starts_with("age",ignore.case = F))) %>%
dplyr::select(PID,Sample_ID,Sample_Type,Age,Gender,Stage,everything())
tmp_data[tmp_data == ""] <- NA
}else{
tmp_data <- tibble::column_to_rownames(var = "Ensembl_ID")
}
saveRDS(tmp_data,paste(destdir,outname,sep = "/"))
cat("File has been downloaded and stored in ",destdir,"\n")
# 删除原始文件
if(RMraw){
file.remove(tmp_file)
}
}数据挖掘中数据以及代码的撰写原则

四、R语言可视化——ggplot2
几个基本概念
- 图层(layer)
- 数据(Data)和映射(Mapping)
- 标度(Scale)
- 几何对象(Geometric)
- 统计变换(Statistics)
- 坐标系统(Coordinate)
- 分面(Facet)

图层和映射


几何对象

- 映射关系既可以放在最底层的图层,也可以放在每一个几何对象的图层,这两者并不冲突
分面

坐标轴

此处纵轴的信息不来自于数据,而是来自于横轴的统计结果
一些标签
练习实例1
> p <- ggplot(data = merged_data, mapping = aes(x=Age, y= OS.time))
> p + geom_point(aes(shape = Gender, color = OS)) + geom_smooth(method = "lm")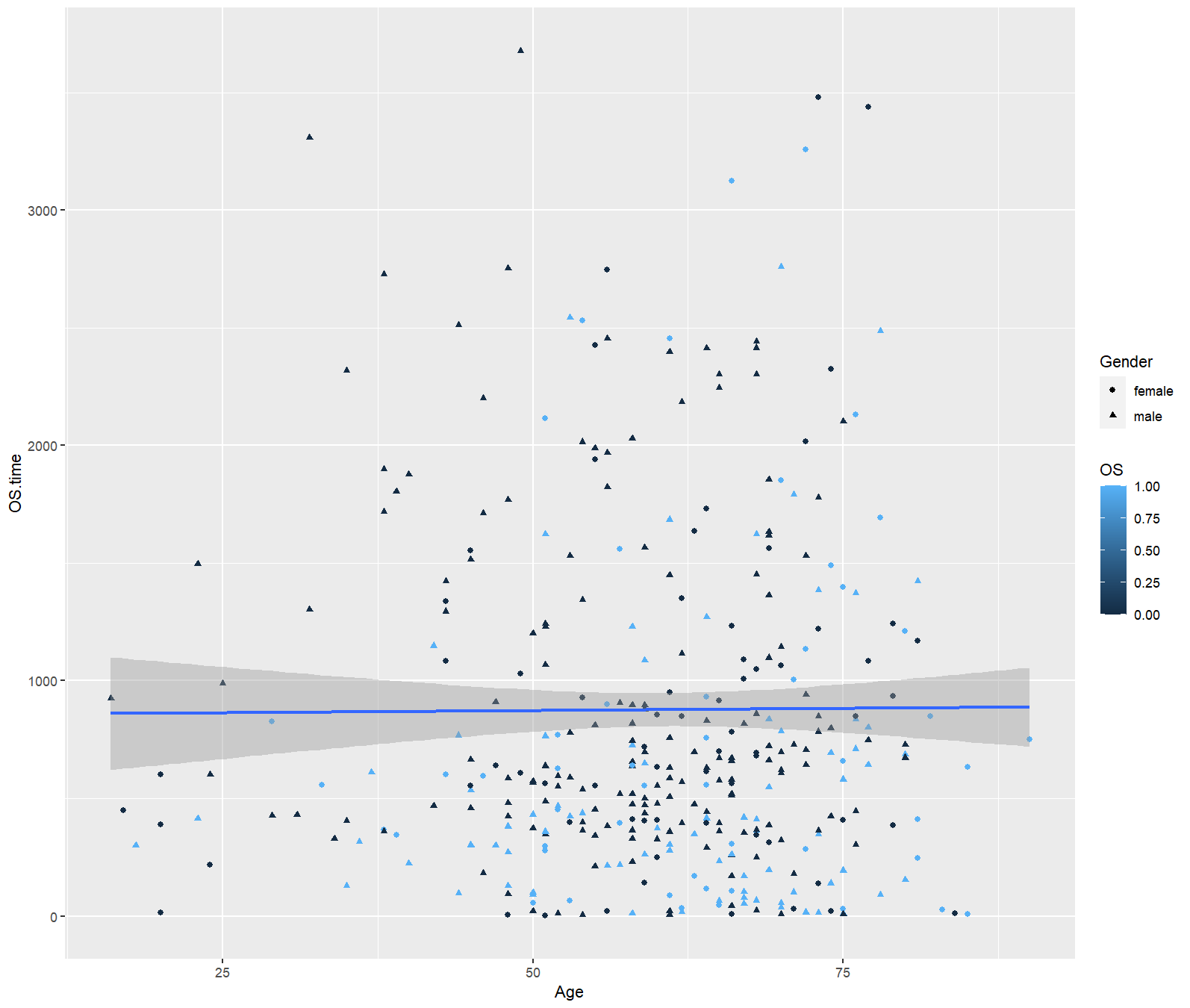
GEO分析以及韦恩图
GEO数据库
关键词:

操作

实例流程
- GEO数据查找以及下载
- 差异基因分析
- 差异基因进行功能富集分析
- 差异基因进行KEGG分析
- 差异基因进行PPI分析
- 筛选Hub基因
- DEGs(differentially expressed genes)在两组不同状态样本中表达量有差异的基因
- GEO或者TCGA均可以拿到基因的表达谱信息用于计算DEGs
- GEO中分为两类数据,一种是芯片数据(在线分析),一种是高通量测序数据(R语言分析)
代码复现

# 检查相应的package是否完成安装
if(!require(dplyr)){install.packages("dplyr")}
if(!require(openxlsx)){install.packages("openxlsx")}
if(!require(data.table)){install.packages("data.table")}
if(!require(VennDiagram)){install.packages("VennDiagram")}
library(dplyr)
library(openxlsx)
library(VennDiagram)
#定义两组对比的结果文件名
sam1 <- "./ReadData/Normal_vs_Cirrhosis.txt"
sam2 <- "./ReadData/Normal_vs_HCC.txt"
outVenn <- "HCC_Cirrhosis.png"
outXlsx <- "./Out/GSE14323_HCC_Cirrhosis.xlsx"
fc.cutoff <- 2
fdr.cutoff <- 0.05
#1. 分别读入并进行简单的过滤,实际分析中需要增强R语言技能进行深度的基因过滤
#包括1. 调整logFC阈值;2. 调整adj.p阈值;3. 没有特定意义的基因滤除(例如LOC开头的基因MIR/MT-开头的基因等)
tb.1 <- data.table::fread(sam1) %>% as.data.frame() %>%
filter(Gene.symbol != "") %>%
mutate(Disease = ifelse(logFC > 0, "Up","Down")) %>%
filter(adj.P.Val <= fdr.cutoff) %>%
filter(logFC >= log2(fc.cutoff) | logFC <= -log2(fc.cutoff)) %>%
filter(!grepl("^MT-|^LOC",Gene.symbol)) %>%
mutate(Label = paste(Gene.symbol,Disease,sep = "_"))
tb.2 <- data.table::fread(sam2) %>% as.data.frame() %>%
filter(Gene.symbol != "") %>%
mutate(Disease = ifelse(logFC > 0, "Up","Down")) %>%
filter(adj.P.Val <= fdr.cutoff) %>%
filter(logFC >= log2(fc.cutoff) | logFC <= -log2(fc.cutoff)) %>%
filter(!grepl("^MT-|^LOC",Gene.symbol)) %>%
mutate(Label = paste(Gene.symbol,Disease,sep = "_"))
#2. 同高或者同低的重复基因的只保留一个
tb.1 <- tb.1 %>% distinct(Label,.keep_all = T)
tb.2 <- tb.2 %>% distinct(Label,.keep_all = T)
#3. 一个基因既高表达又低表达直接剔除
dup1 <- unique(tb.1$Gene.symbol[duplicated(tb.1$Gene.symbol)])
dup2 <- unique(tb.2$Gene.symbol[duplicated(tb.2$Gene.symbol)])
tb.1 <- tb.1 %>% filter(!Gene.symbol %in% dup1)
tb.2 <- tb.2 %>% filter(!Gene.symbol %in% dup2)
#4. 计算两者的overlap并画韦恩图
g1up <- tb.1$Gene.symbol[tb.1$Disease == "Up"]
g1down <- tb.1$Gene.symbol[tb.1$Disease == "Down"]
g2up <- tb.2$Gene.symbol[tb.2$Disease == "Up"]
g2down <- tb.2$Gene.symbol[tb.2$Disease == "Down"]
venn.diagram(x= list(G1up = g1up,G1down = g1down,G2up = g2up,G2down = g2down),
filename = outVenn, height = 450, width = 450,resolution =300,
imagetype="png", col ="transparent",
fill =c("cornflowerblue","green","yellow","darkorchid1"),
alpha = 0.5,
cex = 0.45,fontface = "bold",
cat.col =c("darkblue", "darkgreen", "orange","darkorchid4"),
cat.cex = 0.45, cat.dist = 0.07)
#5. 写出到excel中
bothUp <- intersect(tb.1$Gene.symbol[tb.1$Disease == "Up"],
tb.2$Gene.symbol[tb.2$Disease == "Up"])
bothDown <- intersect(tb.1$Gene.symbol[tb.1$Disease == "Down"],
tb.2$Gene.symbol[tb.2$Disease == "Down"])
out.data <- list(G1data = tb.1,G2data = tb.2,
BothUP = bothUp,BothDown = bothDown)
write.xlsx(out.data,file = outXlsx)TCGA分析以及Bar图,小提琴图,盒装图
TCGA相关数据库以及数据下载
Bar图
概念:bar图也叫做条形图。通常用来展示不同分组之间数量或者百分比的对比,因此只适用于分组变量,如果是连续变量则需要人卫进行分组
实例分析
下载并清洗数据
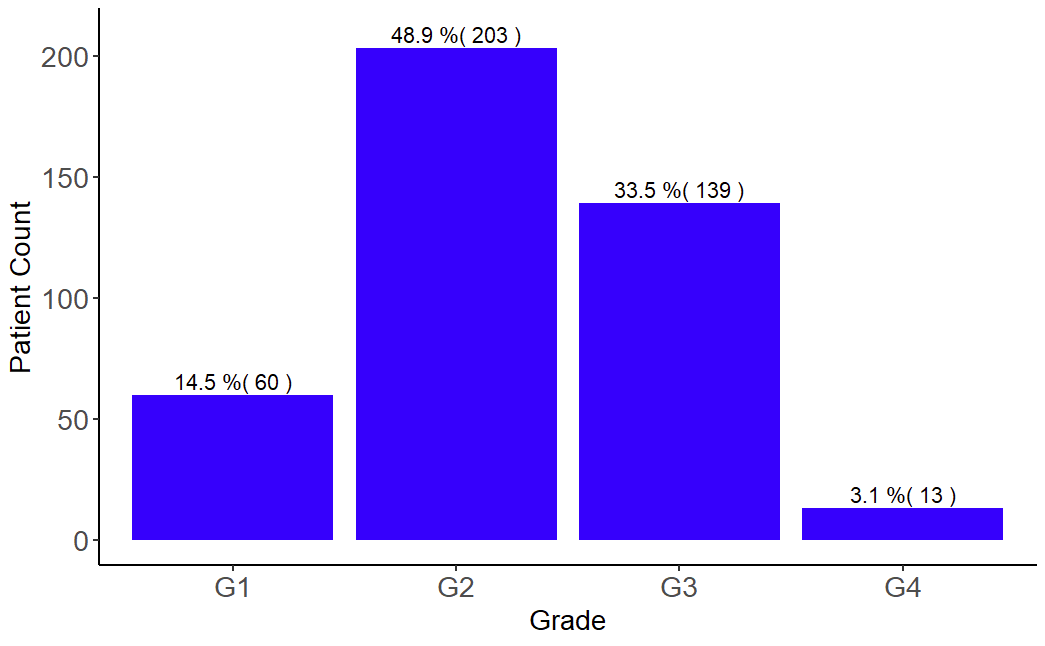
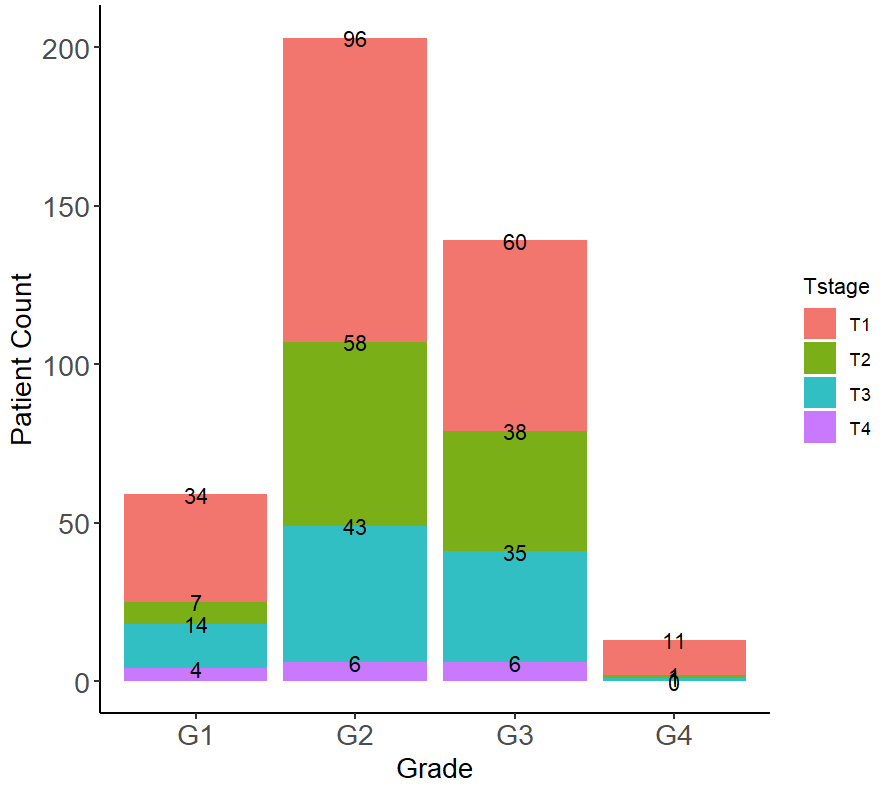
# 下载肝癌中RNA表达谱以及临床数据,然后任意挑选其中一个基因将数据进行合并 plot15_data <- readRDS("./ReadData/TCGA_example.RDS") View(plot15_data) # 提取练习的数据并清洗 plot_raw <- plot15_data %>% select(Grade,Tstage) %>% mutate(Grade = as.character(Grade), Tstage = as.character(Tstage)) %>% filter(grepl("G",Grade))
绘图
# 不同分级的患者比例-1 ggplot(plot_raw,aes(x = Grade)) + geom_bar() # 不同分级的患者比例-1,修改图的细节 plot_data <- data.frame(table(plot_raw$Grade)) %>% mutate(Total = nrow(plot_raw)) %>% mutate(Percentage = round(Freq/Total,3) * 100) ggplot(plot_data,aes(x = Var1, y = Freq)) + geom_bar(stat = "identity",position = "dodge",fill = "blue") + theme_classic() + xlab("Grade") + ylab("Patient Count") + theme(legend.position = "none", axis.text = element_text(size = 14), axis.title = element_text(size = 14)) + geom_text(aes(y = Freq + 6,label = paste(Percentage,"%(",Freq,")"))) #不同分级的患者中T分期的分布 plot_data <- data.frame(table(plot_raw$Grade,plot_raw$Tstage)) ggplot(plot_data,aes(x = Var1, y = Freq, fill = Var2)) + geom_bar(stat = "identity", position = "stack") + labs(x = "Grade", y = "Patient Count", fill = "Tstage") + theme_classic() + theme(axis.text = element_text(size = 14), axis.title = element_text(size = 14)) + geom_text(aes(label = Freq),position = position_stack())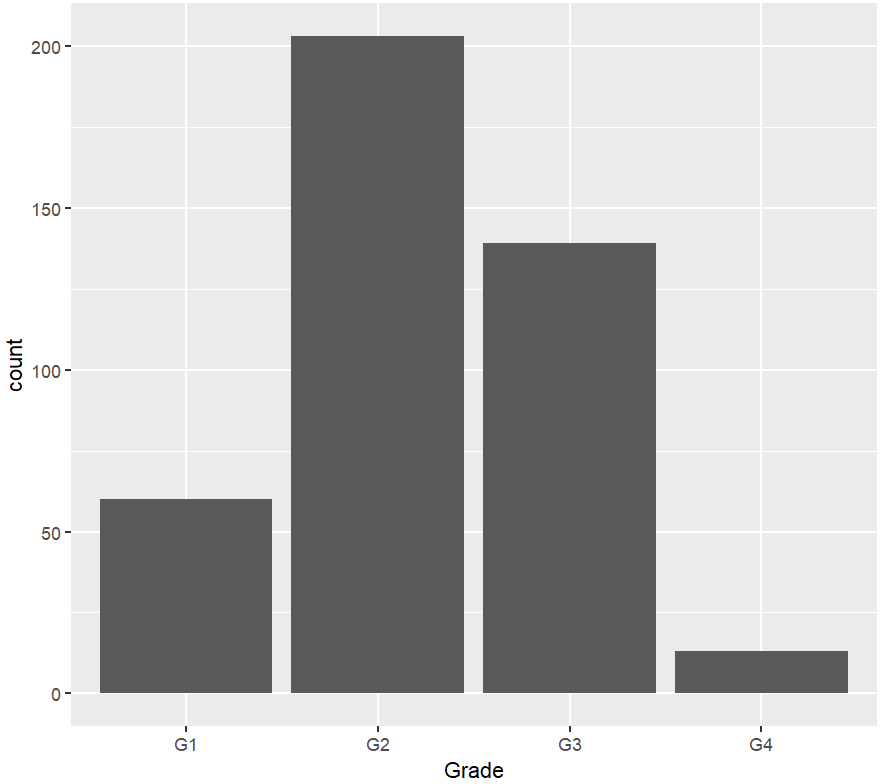
拓展小练习:
plot_dt <- data.frame(table(plot_david$Tstage, plot_david$Gender))
plot_dt$Total <- apply(plot_dt,1,function(x)sum(plot_dt$Freq[plot_dt$Var1 == x[1]]))
plot_dt <- plot_dt %>% mutate(Percentage = round(Freq/Total,3) * 100)
ggplot(plot_dt,aes(x = Var1, y = Percentage, fill = Var2)) +
geom_bar(stat = "identity", position = "stack") +
labs(x = "Grade", y = "Percentage(%)", fill = "Tstage") +
theme_classic() +
theme(axis.text = element_text(size = 14),
axis.title = element_text(size = 14)) +
geom_text(aes(label = Freq),position = position_stack()) +
scale_fill_jco()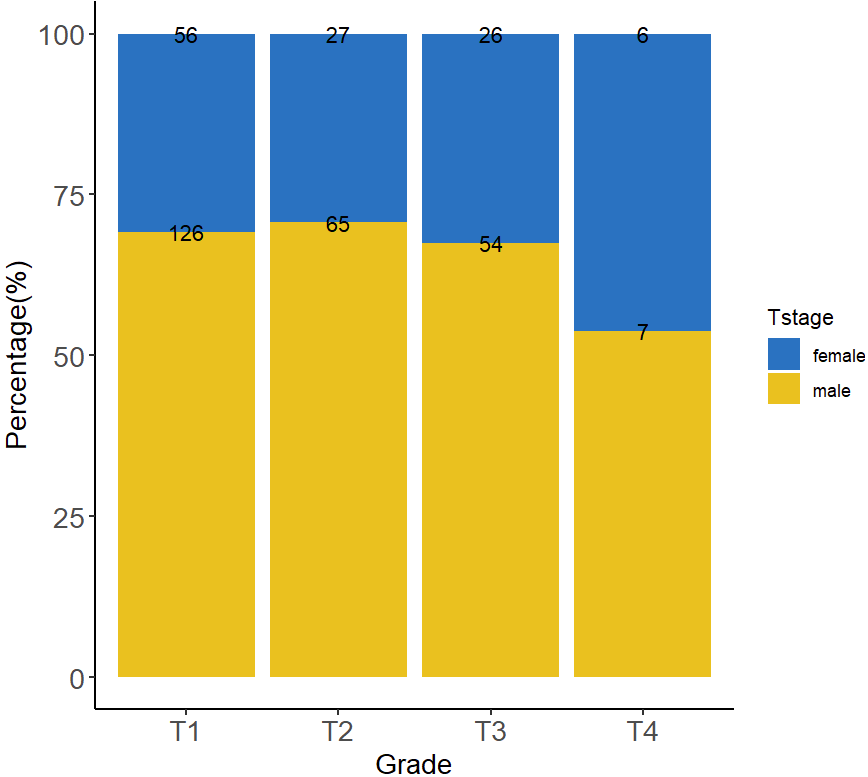
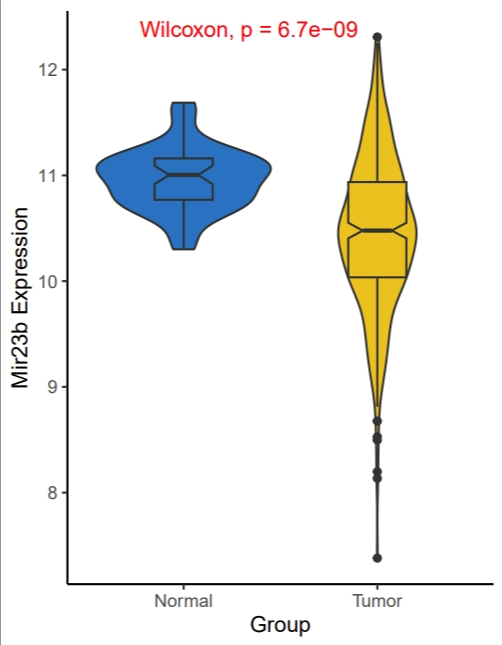
盒状图以及小提琴图(箱形图)
盒状图:
- 是一种用作显示一组数据分散情况的统计图,因形状如箱子而得名;主要用于反映原始数据分布的特征,并且可以进行多组数据分布特征的比较
- 箱形图能够显示出一组数据的最大值、最小值、中位数以及上下四分位数,同时还可以显示逸出值

实例分析
#不同类型样本(肿瘤以及癌旁)中某基因表达量的对比图
plot_data <- plot15_data %>% select(Group,Mir23b)
p <- ggplot(plot_data,aes(x = Group,y = Mir23b, fill = Group))+
geom_boxplot(notch = T) +
theme_classic() +
ggpubr::stat_compare_means(color = "red") +
ggsci::scale_fill_jco() +
theme(legend.position = "none") +
ylab("Mir23b Expression")
ggsave(filename = "./Out/Group_Mir23b_box.pdf",plot = p,
width = 3.5,height = 4.5)
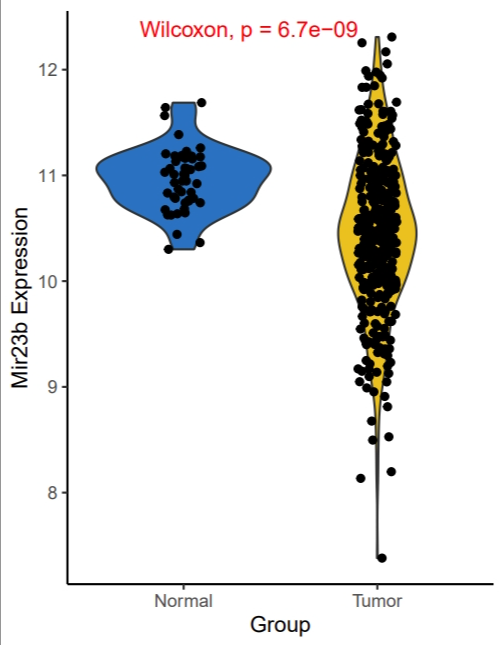
p <- ggplot(plot_data,aes(x = Group,y = Mir23b, fill = Group))+
geom_violin() + geom_boxplot(width = .3,notch = T) +
theme_classic() +
ggpubr::stat_compare_means(color = "red") +
ggsci::scale_fill_jco() +
theme(legend.position = "none") +
ylab("Mir23b Expression")
ggsave(filename = "./Out/Group_Mir23b_violin.pdf",plot = p,
width = 3.5,height = 4.5)
p <- ggplot(plot_data,aes(x = Group,y = Mir23b, fill = Group))+
geom_violin() +
geom_jitter(width = .1) +
theme_classic() +
ggpubr::stat_compare_means(color = "red") +
ggsci::scale_fill_jco() +
theme(legend.position = "none") +
ylab("Mir23b Expression")
ggsave(filename = "./Out/Group_Mir23b_jitter.pdf",plot = p,
width = 3.5,height = 4.5)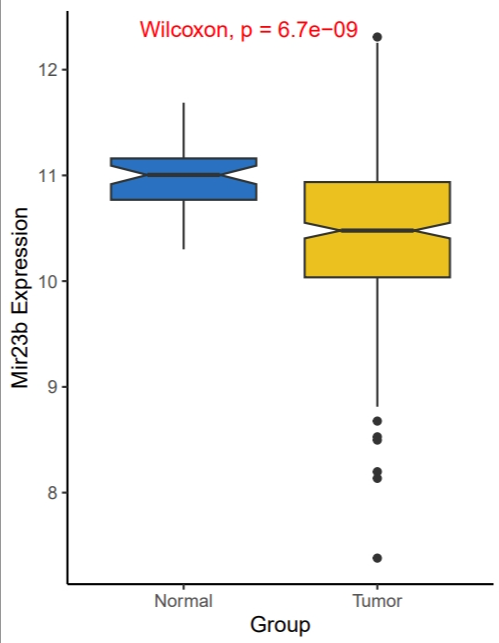
差异表达基因分析以及火山图
一些小知识点
差异基因分析-非count数据(normalize之后的数据)
- limma:芯片测序数据(array——>GEO2R)
- wilcox-test:FPKM/TOM等标准化数据
差异基因分析-count数据(推荐使用)
- DESeq2:高通量测序count
- limma-voom:高通量测序count
非count数据进行差异分析
- 芯片数据(array):最简单直接使用在线工具GEO2R即可
高通量中的非count数据:
- FPKM(XENA-TCGA提供的为此种数据)

- RPKM
- CPM
- TPM(当前比较推荐的标准化数据,transcript per million)
- FPKM(XENA-TCGA提供的为此种数据)
XENA中下载到的TCGA数据基因需要更换到official symbol,先计算再变更或者先变更再计算都可以,使用代码如下:
library(org.Hs.eg.db)
library(AnnotationDbi)
lumb_res$Gene <- mapIds(org.Hs.eg.db,
keys = as.character(lumb_res$Ensembl),
column = "SYMBOL",
keytype = "ENSEMBL",
multiVals = "first")TCGA数据或者GEO数据中的FPKM/TPM数据处理
- 读入表达矩阵并转换成FPKM数据
- 读入临床数据并提取样本分组信息
- 调用写好的函数直接计算差异表达基因
GEO中的芯片数据处理
- 查找数据,切换到GEO2R
- 整理样本分组并计算
- 导出计算结果
TCGA-RNA数据计算DEGs实例(非count数据)
#读入临床信息提取差异分析的样本分组
> tcga_clic_raw <- data.table::fread("./ReadData/TCGA-LIHC.GDC_phenotype.tsv.gz")
> tcga_clic <- tcga_clic_raw %>% as.data.frame() %>%
+ dplyr::select(submitter_id.samples,
+ age_at_initial_pathologic_diagnosis,
+ pathologic_M,
+ sample_type.samples) %>%
+ dplyr::rename(Sample_ID = submitter_id.samples,
+ Age = age_at_initial_pathologic_diagnosis,
+ Mstage = pathologic_M,
+ SamType = sample_type.samples)
#清洗数据
> tcga_clic <- tcga_clic %>%
+ mutate(SamType = case_when(
+ grepl("Primary",SamType) ~ "Primary",
+ grepl("Recurrent",SamType) ~ "Recurrent",
+ grepl("Normal",SamType) ~ "Normal"
+ ))
#读入表达矩阵
> tcga_expr <- data.table::fread("./ReadData/TCGA-LIHC.htseq_fpkm.tsv.gz") %>% as.data.frame()
#清洗数据
> tcga_expr$Ensembl_ID <-
+ sub("(ENSG\\d+)\\..*","\\1",tcga_expr$Ensembl_ID)
> View(tcga_expr)
> tcga_expr$Gene <- mapIds(org.Hs.eg.db,
+ keys = as.character(tcga_expr$Ensembl_ID),
+ column = "SYMBOL",
+ keytype = "EMSEMBL",
+ multiVals = "first")
#转变为可读数据(变为可以分析的表达矩阵)
> tcga_expr <- tcga_expr %>%
+ dplyr::select(-Ensembl_ID) %>%
+ filter(!is.na(Gene)) %>%
+ distinct(Gene,.keep_all = T) %>%
+ column_to_rownames(var = "Gene") %>%
+ as.matrix()
#调用函数计算差异表达基因
> source("./Codes/Wilcox_DEG.R")
> sample_group <- tcga_clic %>%
+ dplyr::select(Sample_ID,SamType) %>%
+ dplyr::rename(Type = SamType) %>%
+ filter(Type %in% c("Normal","Primary"))
> tcga_degs_test <- wilDEGs(expr_matrix =
+ tcga_expr[1:1000,],
+ sample_df = sample_group,
+ pval = 0.05,fdr = 0.05,FC = 2)
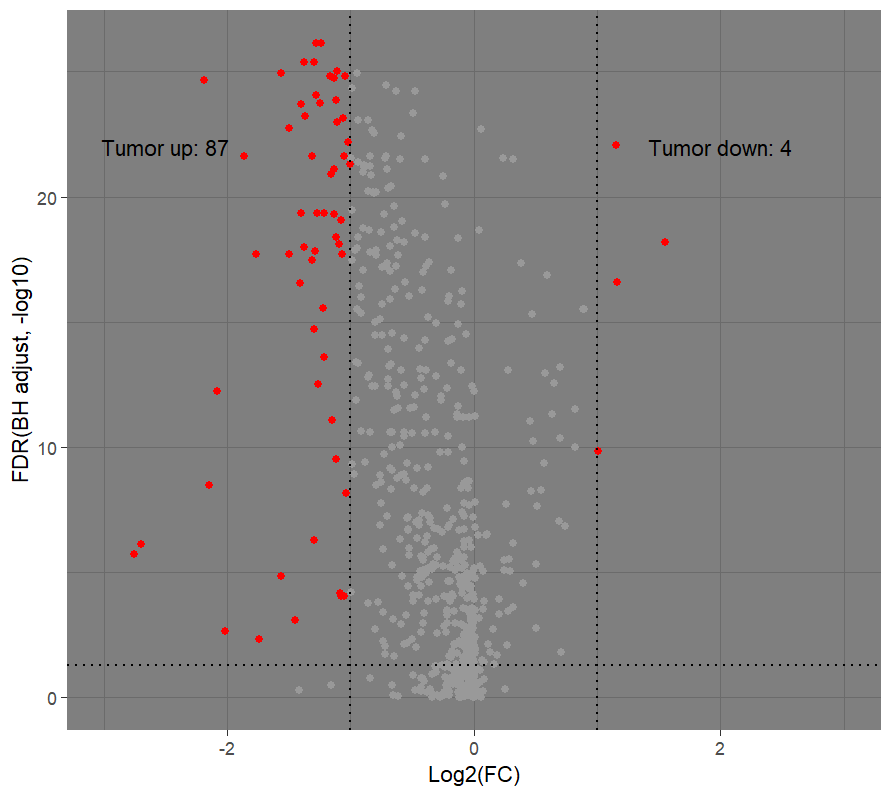
#查看计算结果
> table(tcga_degs_test$Label)
Normal_down Normal_up Stable
60 4 936
#画火山图
plot_data <- tcga_degs_test %>%
mutate(Plog = -log10(FDR)) %>%
mutate(Type = ifelse(Label == "Stable","Stable","Diff"))
ggplot(plot_data,aes(x = LogFC, y = Plog, color = Type)) +
# 画点图
geom_point() +
# 设置背景风格,还可以自己修改成其他风格
theme_bw() +
# 设置x轴的显示范围
xlim(-3,3) +
# 手动修改配色也可以用ggsci::scale_color_aaas()等
scale_color_manual(values = c("red","grey60")) +
# 画一条横线两条竖线,linetype后面的数字可以控制线类型
geom_hline(yintercept =-log10(0.05),linetype = 3) +
geom_vline(xintercept =c(-1,1),linetype = 3) +
# 把图例去掉
theme(legend.position = "none") +
# 增加两句文字在图中丰富信息使用
annotate("text",x = 2,y = 22, label = "Tumor down: 4") +
annotate("text",x = -2.5,y = 22, label = "Tumor up: 87") +
# 修改一下横轴和纵轴的标题,更明确意义
labs(x = "Log2(FC)", y = "FDR(BH adjust, -log10)")最后结果
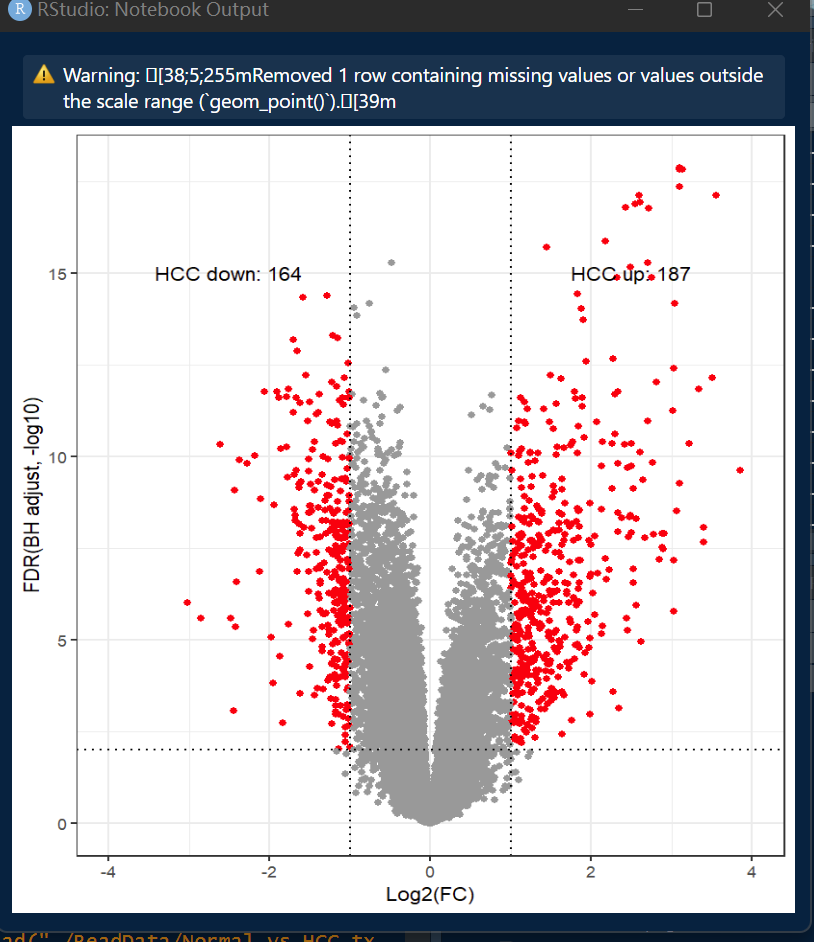
GEO2R数据分析实例
#读入数据
> plot_data <- data.table::fread("./ReadData/Normal_vs_HCC.txt") %>% as.data.frame()
#数据清洗
> plot_data <- plot_data %>%
+ filter(Gene.symbol != "" & !is.na(Gene.symbol)) %>%
+ dplyr::rename(LogFC = logFC) %>%
+ mutate(Plog = -log10(adj.P.Val)) %>%
+ mutate(Type = ifelse(abs(LogFC) > 1 & adj.P.Val < 0.01,"Diff","Stable"))
#ggplot绘图
> ggplot(plot_data,aes(x = LogFC, y = Plog, color = Type)) +
+ geom_point() +
+ theme_bw() +
+ xlim(-4,4) +
+ scale_color_manual(values = c("red","grey60")) +
+ geom_hline(yintercept =-log10(0.01),linetype = 3) +
+ geom_vline(xintercept =c(-1,1),linetype = 3) +
+ theme(legend.position = "none") +
+ annotate("text",x = 2.5,y = 15, label = "HCC up: 187") +
+ annotate("text",x = -2.5,y = 15, label = "HCC down: 164") +
+ labs(x = "Log2(FC)", y = "FDR(BH adjust, -log10)")
最后结果:
相关性点图分析
聚类分析以及热图
热图:展示多组之间的特征对比
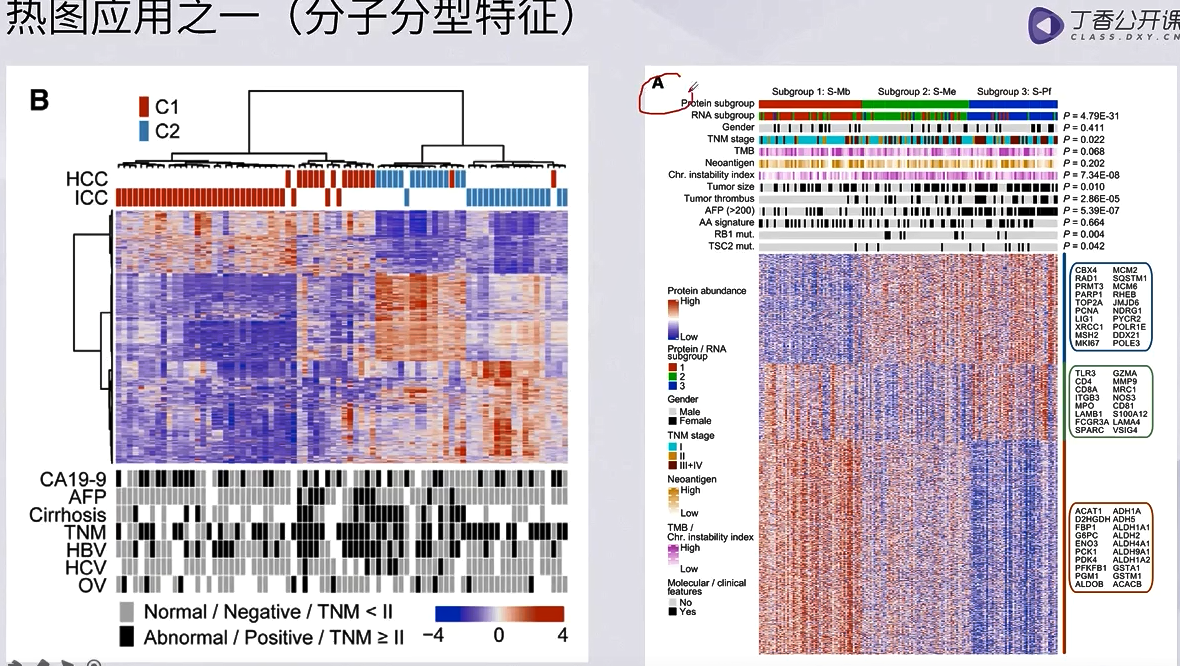

画热图的tips
要先进行z-score的转换
- 热图的包:ComplexHeatmap
#读取数据
> plot_heatmap <- readRDS("./ReadData/HeatmapData.RDS")
> plot_cluster <- readRDS("./ReadData/Cluster_id.RDS")
#热图自动聚类
col_fun = circlize::colorRamp2(c(-2,-1, 0, 1.2, 2),
c("cornflowerblue","deepskyblue1", "white","red2", "red"))#选择颜色
Heatmap(plot_heatmap,#画图的数据
col = col_fun,#颜色
column_km = 2,#把列聚成几类?
row_names_side = c("left"),#呈现行名在左边
show_row_dend = T,#聚类树显示
show_column_dend = T,#聚类树显示
cluster_columns = T,#列是否做聚类
cluster_rows = T,#行是否做聚类
row_names_gp = gpar(fontsize = 6),#行名调整
heatmap_legend_param = list(title = "Z-score"),
show_column_names = T)#显示列名")
#热图增加注释
> col_fun = circlize::colorRamp2(c(-2,-1, 0, 1.2, 2), c("tan4","tan", "white","red2", "red"))
> ht1 = HeatmapAnnotation(Cluster = plot_cluster$Cluster,
+ show_legend = F,annotation_name_side = "left",
+ col = list(Cluster = c("Type1"="#BC3C29FF",
+ "Type2"="#0072B5FF",
+ "Type3"="#E18727FF",
+ "Type4"="#20854EFF")))
> Heatmap(plot_heatmap,
+ col = col_fun,
+ #column_km = 2,
+ column_split = plot_cluster$Cluster,
+ top_annotation = ht1,
+ row_names_side = c("left"),
+ show_row_dend = F,
+ show_column_dend = F,
+ cluster_columns = T,
+ cluster_rows = F,
+ row_names_gp = gpar(fontsize = 6),
+ heatmap_legend_param = list(title = "Z-score"),
+ show_column_names = T)
富集分析以及条形图气泡图GSEA
常见的富集分析类型:GO/KEFF/GSEA
GO:gene ontology,主要记录基因的具体功能分类
- BP-biology process
- MF-molecu function
- CC-cellular component
- Pathway:主要指KEGG等pathway,记录基因上下游的调控关系以及相关构成的通路网络
- 在线分析数据库:DAVID, Enrich, WebGenestalt等
- R语言分析包:clusterProfiler
实例分析(GO富集)
##数据清洗
#读入数据
> tcga_clic_raw <- data.table::fread("./ReadData/TCGA-LIHC.GDC_phenotype.tsv.gz")
> tcga_clic <- tcga_clic_raw %>% as.data.frame() %>%
+ dplyr::select(submitter_id.samples,
+ age_at_initial_pathologic_diagnosis,
+ pathologic_M,sample_type.samples) %>%
+ dplyr::rename(Sample_ID = submitter_id.samples,
+ Age = age_at_initial_pathologic_diagnosis,
+ Mstage = pathologic_M,
+ SamType = sample_type.samples)
#清洗数据
> tcga_clic <- tcga_clic %>%
+ mutate(SamType = case_when(
+ grepl("Primary",SamType) ~ "Primary",
+ grepl("Recurrent",SamType) ~ "Recurrent",
+ grepl("Normal",SamType) ~ "Normal"
+ ))
#读入表达矩阵
> tcga_expr <- data.table::fread("./ReadData/TCGA-LIHC.htseq_fpkm.tsv.gz") %>% as.data.frame()
#清洗数据
> tcga_expr$Ensembl_ID <- sub("(ENSG\\d+)\\..*","\\1",tcga_expr$Ensembl_ID)
> tcga_expr$Gene <- mapIds(x = org.Hs.eg.db,
+ keys = tcga_expr$Ensembl_ID,
+ column = "SYMBOL",
+ keytype = "ENSEMBL",
+ multiVals = "first")
#转换成为可以分析的表达矩阵
> tcga_expr <- tcga_expr %>%
+ dplyr::select(-Ensembl_ID) %>%
+ filter(!is.na(Gene)) %>%
+ distinct(Gene,.keep_all = T) %>%
+ column_to_rownames(var = "Gene") %>%
+ as.matrix()
> tcga_expr <- 2^tcga_expr - 1
##依据年龄计算差异表达基因
> sample_group <- tcga_clic %>%
+ filter(SamType == "Primary",!is.na(Age)) %>%
+ mutate(Age = ifelse(Age >= median(Age),"Old","Young")) %>%
+ dplyr::select(Sample_ID,Age) %>%
+ dplyr::rename(Type = Age)
> table(sample_group$Type)
Old Young
196 180
> tcga_degs_test <- wilDEGs(expr_matrix = tcga_expr,
+ sample_df = sample_group,
+ pval = 0.01,fdr = 0.5,FC = 1.5)
Shared sample count 370
Finished cal of mean Young
Finished cal of mean Old
Finished cal of Pvalue
> table(tcga_degs_test$Label)
Old_down Old_up Stable
394 258 32802
# 筛选出来差异表达的基因
> diff_genes <- tcga_degs_test %>%
+ filter(Label != "Stable") %>%
+ filter(!is.infinite(LogFC))
##GO富集分析
> entrzid = bitr(diff_genes$Gene,
+ fromType = "SYMBOL",
+ toType = "ENTREZID",
+ OrgDb = "org.Hs.eg.db")
> ego_MF <- enrichGO(OrgDb = "org.Hs.eg.db",
+ keyType = "ENTREZID",
+ gene = entrzid$ENTREZID,
+ qvalueCutoff = 0.05,
+ ont = "MF",
+ readable = TRUE)
> ego_CC <- enrichGO(OrgDb = "org.Hs.eg.db",
+ keyType = "ENTREZID",
+ gene = entrzid$ENTREZID,
+ qvalueCutoff = 0.05,
+ ont = "CC",
+ readable = TRUE)
> ego_BP <- enrichGO(OrgDb = "org.Hs.eg.db",
+ keyType = "ENTREZID",
+ gene = entrzid$ENTREZID,
+ qvalueCutoff = 0.05,
+ ont = "BP",
+ readable = TRUE)
> ego_ALL <- enrichGO(OrgDb = "org.Hs.eg.db",
+ keyType = "ENTREZID",
+ gene = entrzid$ENTREZID,
+ qvalueCutoff = 0.05,
+ ont = "ALL",
+ readable = TRUE)
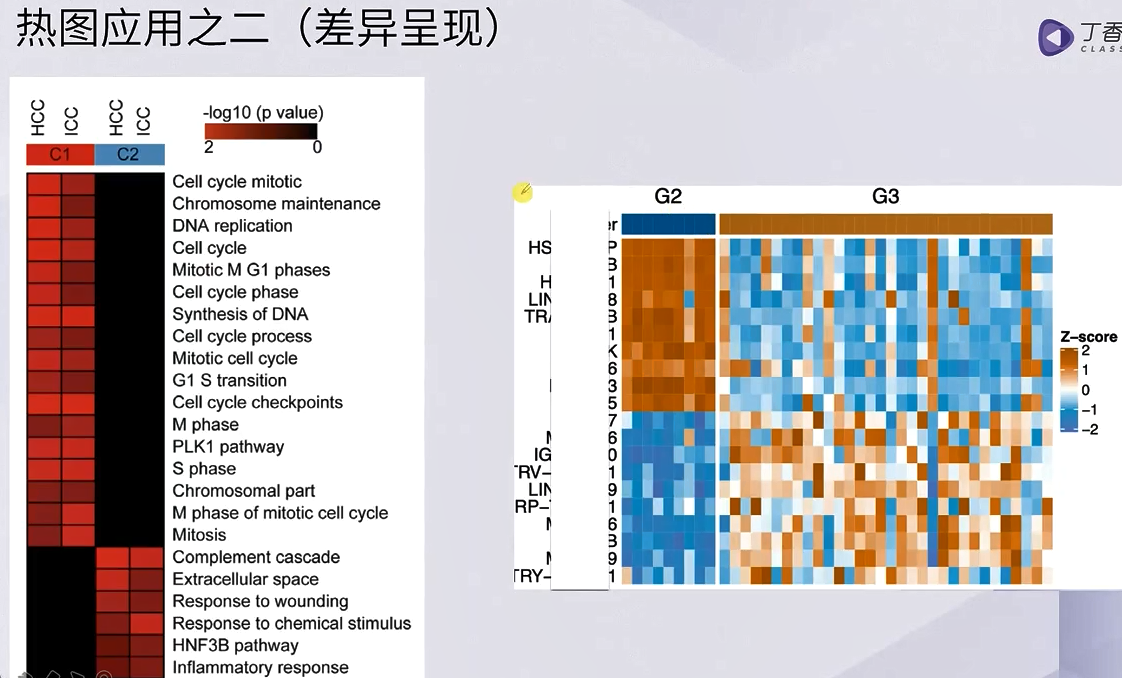
> dotplot(ego_MF)
> barplot(ego_ALL)输出结果: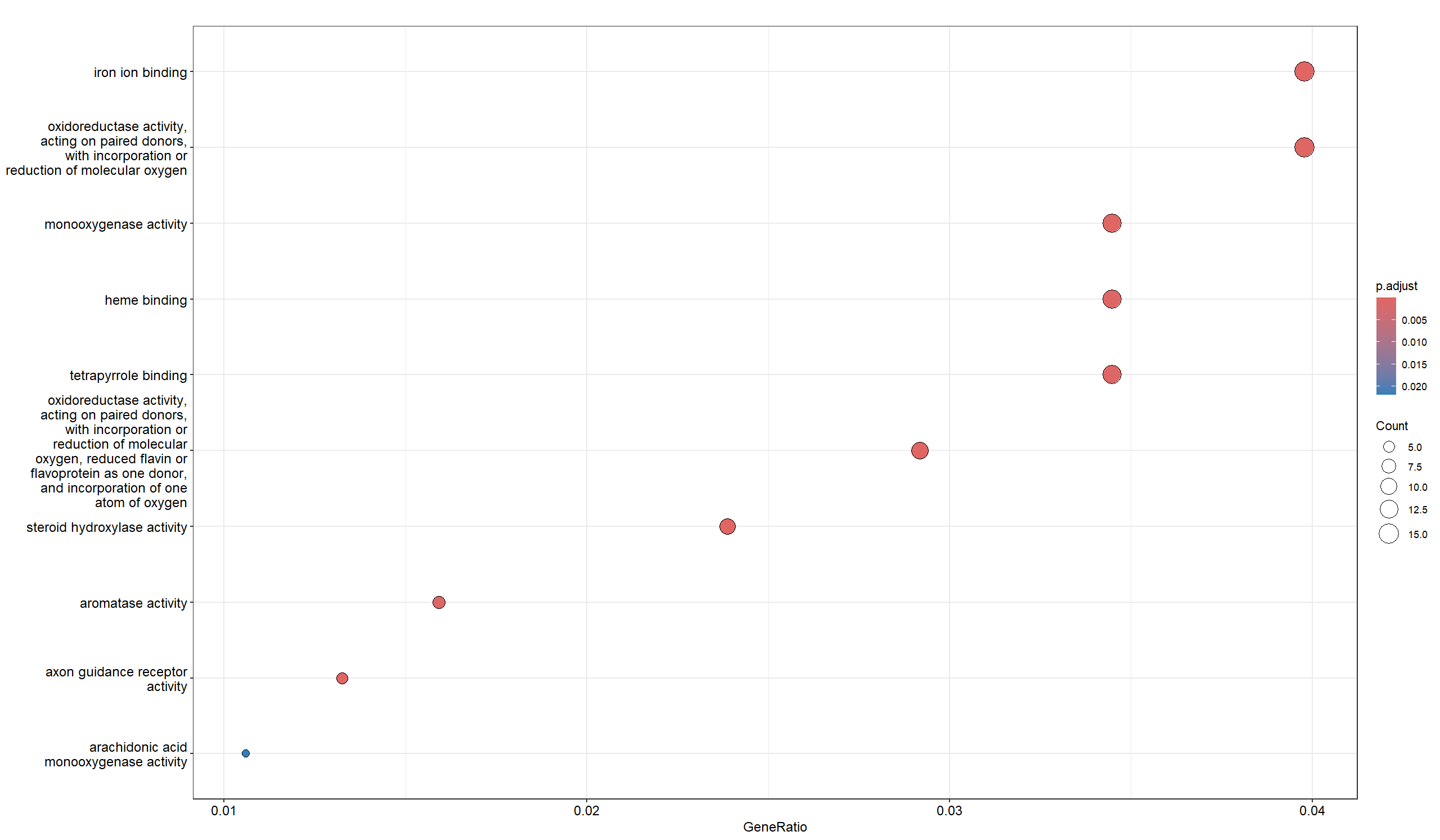
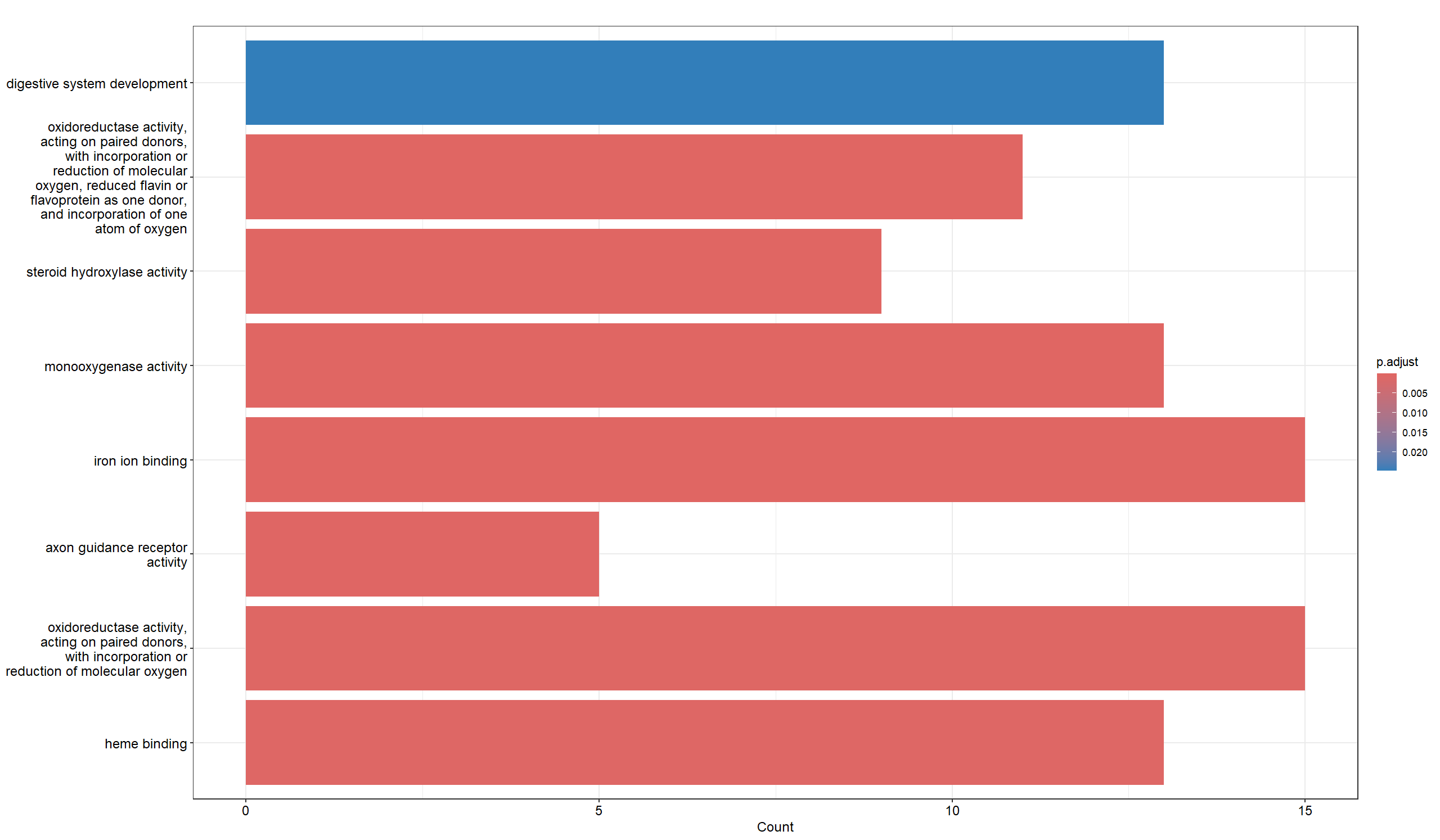
修改例子(使用ggplot2重画图)
代码参考(尚未实际学习)
write.xlsx(ego_MF,file = "./Out/DEGs_go_mf.xlsx")
ego_MF <- read.xlsx("./Out/DEGs_go_mf.xlsx",sheet = 1)
ego_MF <- ego_MF %>% arrange(p.adjust) %>%
mutate(Description = factor(Description,levels = Description,ordered = T))
ggplot(ego_MF[1:6,],
aes(y = Description, x = Count, fill = p.adjust)) +
geom_bar(stat = "identity",position = "dodge")+
scale_fill_gradient(low = "blue",high = "red") +
theme_bw()
ggplot(ego_MF[1:6,],
aes(y = Description, x = Count, color = p.adjust)) +
geom_point(aes(size = Count))+
scale_color_gradient(low = "blue",high = "red") +
theme_bw()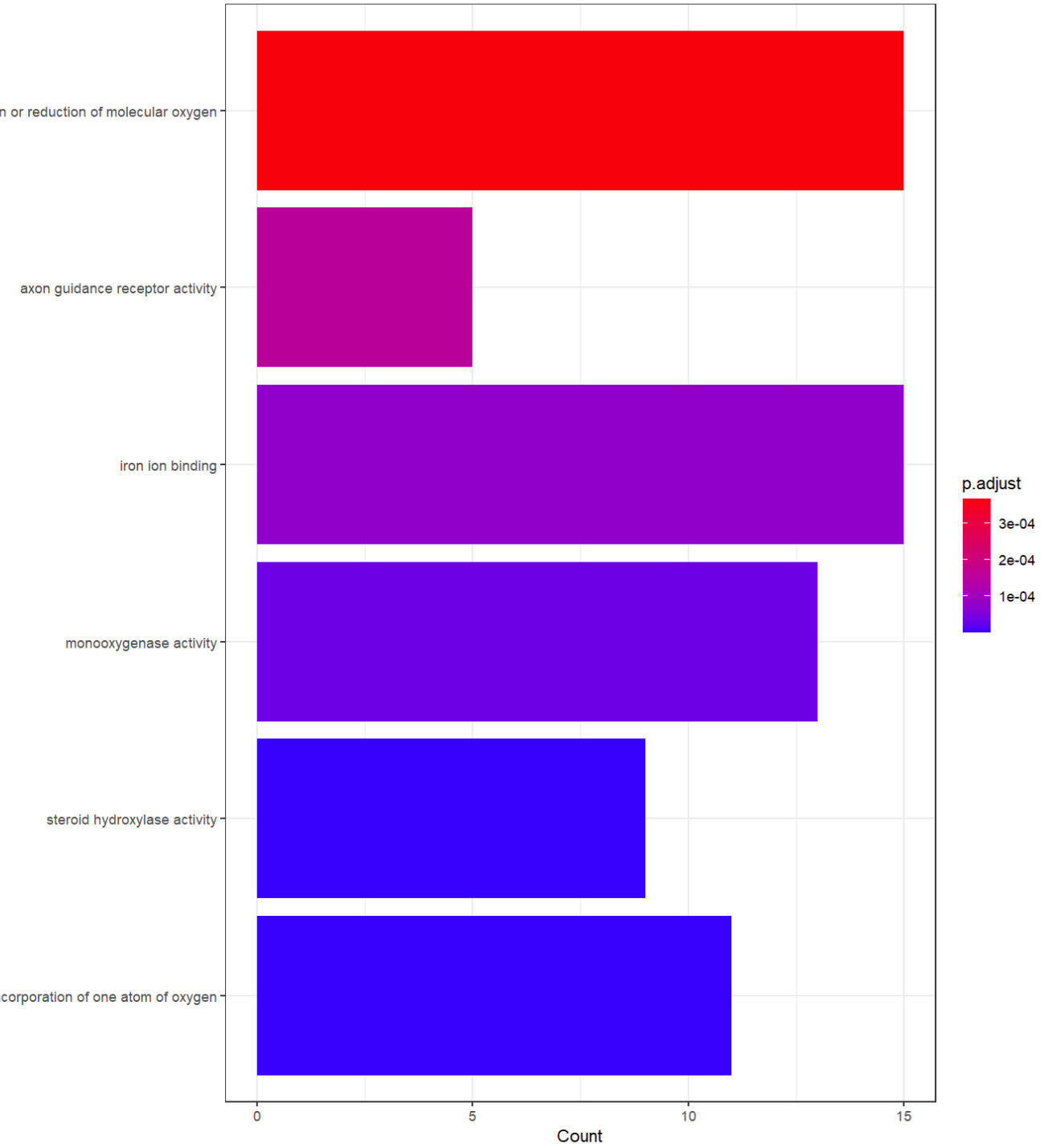
KEGG分析
代码参考
kegg <- enrichKEGG(
gene = entrzid$ENTREZID, #基因列表文件中的基因名称
keyType = 'kegg',
organism = 'hsa',
pAdjustMethod = 'fdr', #指定 p 值校正方法
pvalueCutoff = 0.2, #指定 p 值阈值
qvalueCutoff = 0.2, #指定 q 值阈值
use_internal_data = T
)
dotplot(kegg)
barplot(kegg)GSEA分析
代码参考
# 生成一个带name的向量,向量的值为基因的排序参考,通常使用logFC作为排序的参考依据
entrzid <- entrzid %>%
left_join(diff_genes %>% dplyr::rename(SYMBOL = Gene))
gene_list <- entrzid$LogFC
names(gene_list) <- entrzid$ENTREZID
gene_list <- sort(gene_list,decreasing = T)
# 使用gseGO进行GSEA分析,这里的ont也可以选择不同类型
gseaGO <- gseGO(geneList = gene_list,
ont = 'BP',
OrgDb = "org.Hs.eg.db",
nPerm = 1000,
minGSSize = 10,
pvalueCutoff = 0.2,
verbose = FALSE)
View(as.data.frame(gseaGO))
gseaplot2(gseaGO,
color = "red",
geneSetID = "GO:0032879",
pvalue_table = T)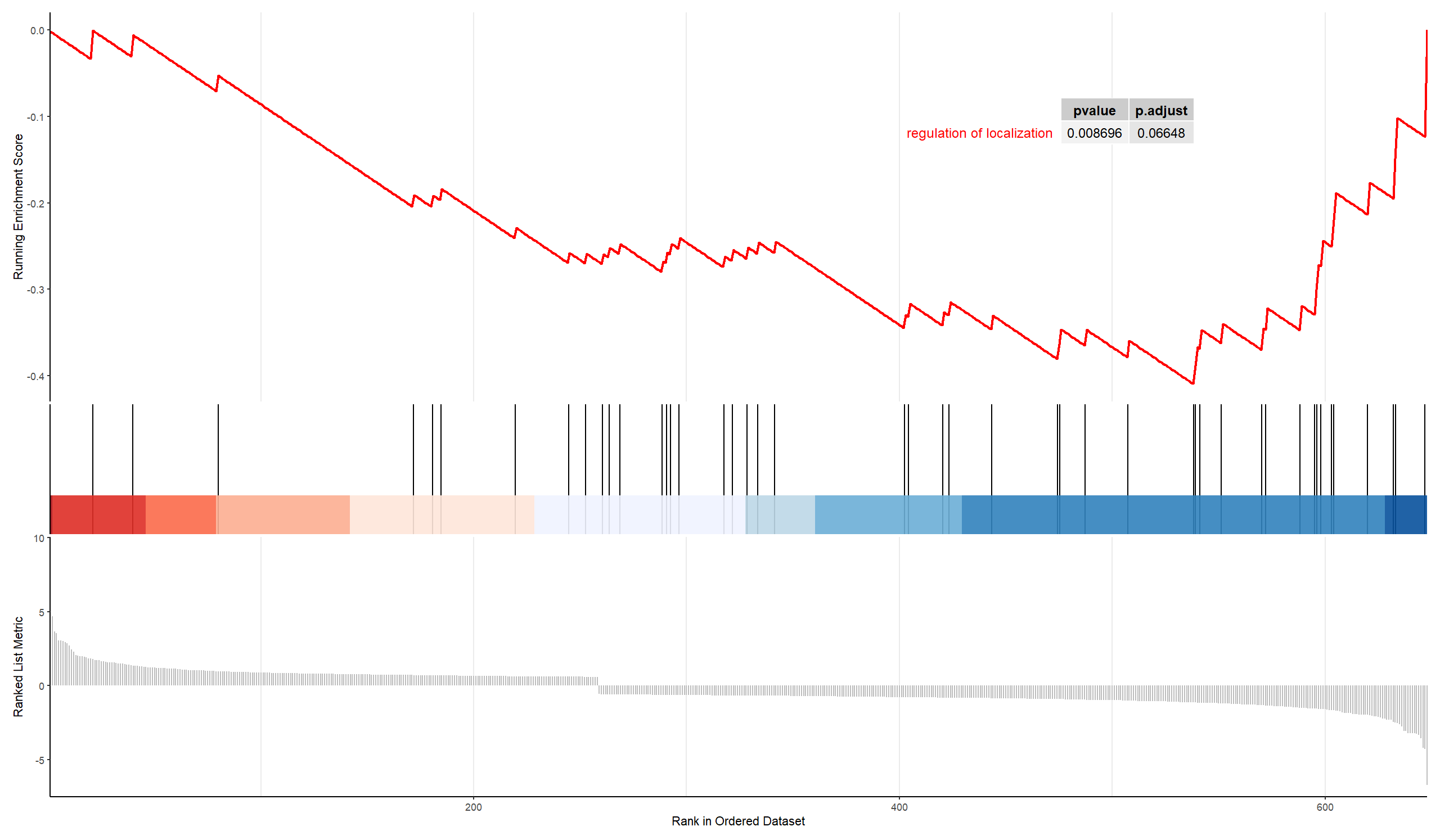
五、R语言实战
生存分析与KM曲线森林图
- 单因素生存分析与作图:KM曲线,cox单因素分析
- 多因素生存分析与作图:森林图
关于生存分析需要知道的
生存数据中应该包含两个部分作为整体
- 生存时间:从入组到最后一次随访的时间间隔
- 生存状态:最后一次随访时患者是否发生了目标事件
- 举例:

- 生存分析会同时考虑以上两个变量,因此不同于常规的分析方法
- R语言中生存分析常用的包时survival(常规分析用), survminer(画图)
单因素生存分析
分类变量:
- log-rank检验
连续变量:
- cox分析
- 转换成分类变量进行log-rank分析
p值计算与HR值计算
- Log-rank可以计算p值
- cox既可以用来计算p值也可以用来计算HR
p值:图中曲线之间是否有显著差异
HR:评估风险
例子分析
#清洗数据
> surv_data <- tcga_clic %>%
+ left_join(tcga_clic_surv %>%
+ dplyr::select(sample,OS,OS.status,Gender,Vascular) %>%
+ dplyr::rename(Sample_ID = sample) %>%
+ unique())
> plot_data <- surv_data %>% filter(SamType == "Primary") %>%
+ mutate(OS = round(OS/30,1))
##一段代码解决单因素分类变量的分析
> fit <- survfit(Surv(OS, OS.status) ~ Mstage, data = plot_data)
#输出图片
> p <- ggsurvplot(fit)
> ggsave(filename = "./Out/Surv_test_1.png",p$plot,width = 4.5,height = 4)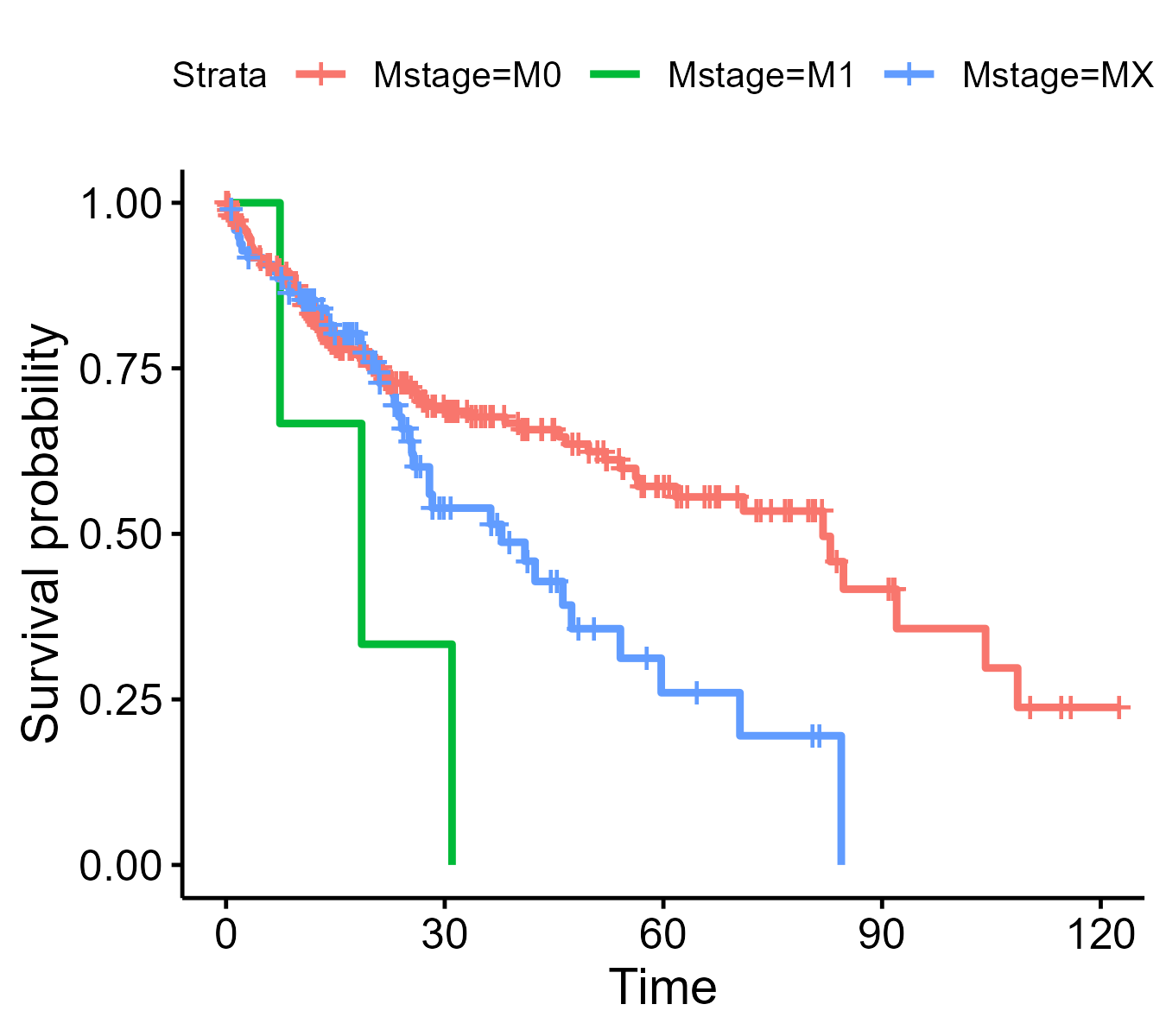
#一些优化
# 增加p值并修改颜色
p <- ggsurvplot(fit,palette = "nejm",pval = T)
ggsave(filename = "./Out/Surv_test_2.png",p$plot,width = 4.5,height = 4)
#增加risktable
> ggsurvplot(fit,palette = "nejm",pval = T,risk.table = T)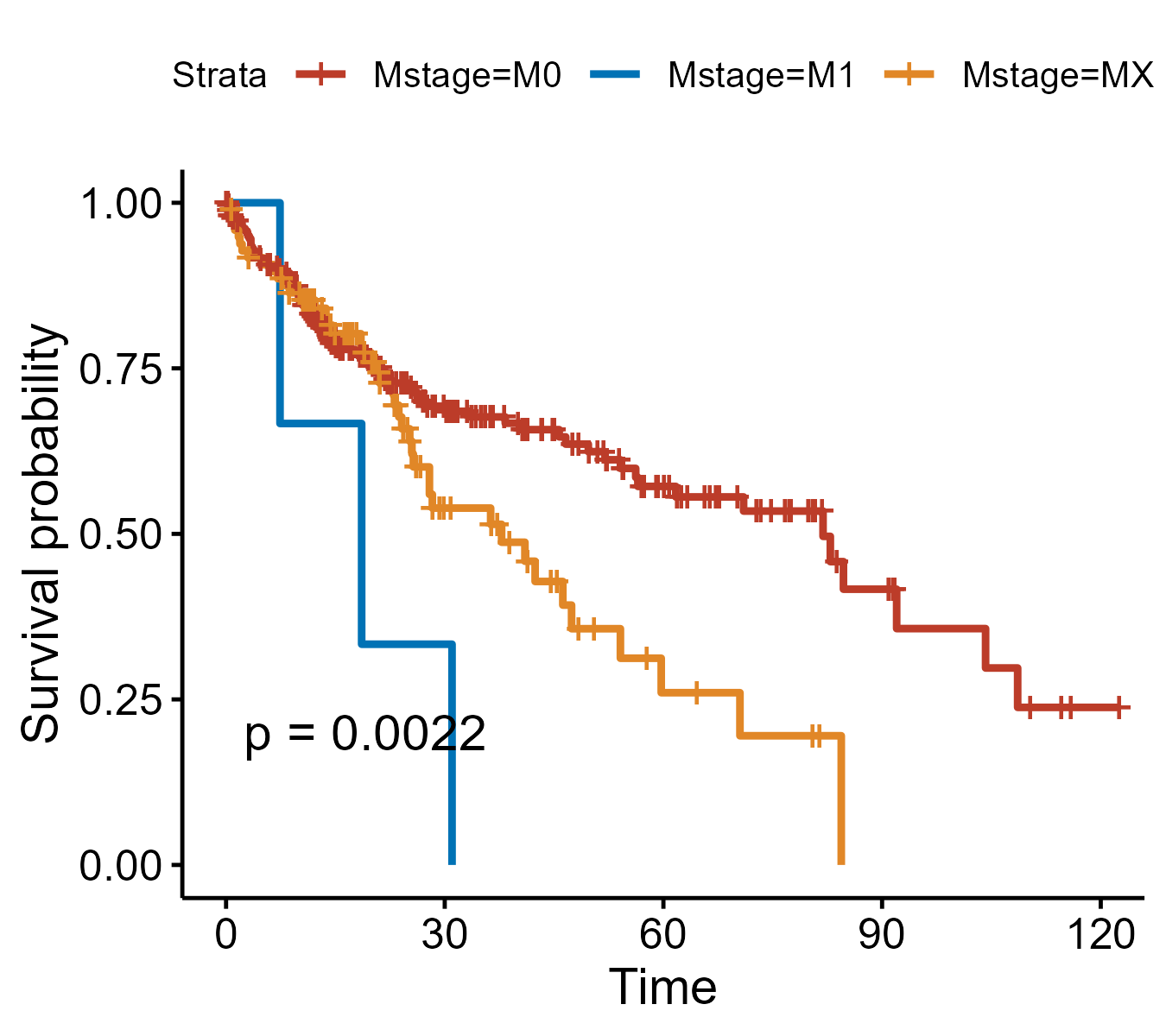
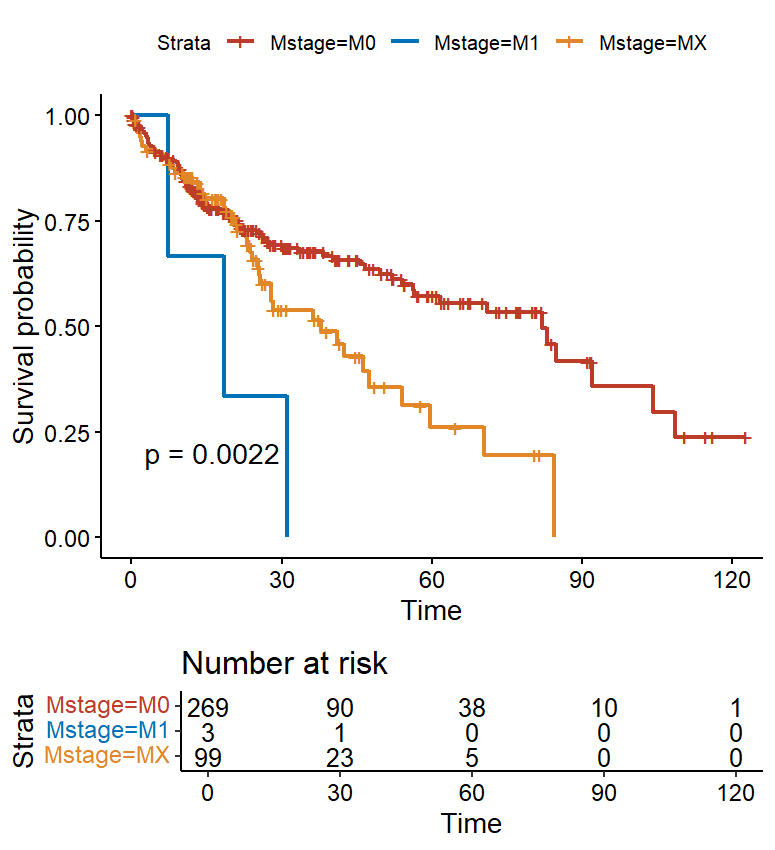
#自动脚本化
source("../Codes/Surv_box_plot.R")
pdf("./Out/Surv_test_4.pdf",width = 4.5,height = 4)
p <- SurvPlot(plot_data = plot_data,
group_label = "Mstage",type = "cat",survival = "OS",label.x = 20)
p$plot
dev.off()cox生存分析(单因素或者多因素)
例子分析
##一、使用分类变量计算
#清洗数据
> plot_data <- plot_data %>% filter(!is.na(Age)) %>%
+ mutate(AgeGroup = ifelse(Age >= median(Age),"Old","Young"))
#分析数据
fit.cox <- coxph(Surv(OS,OS.status) ~ AgeGroup,data = plot_data)
fit.cox.sum <- summary(fit.cox)
# Pvalue
fit.cox.sum$waldtest
fit.cox.sum$logtest
# HR
fit.cox.sum$coefficients[2]
#输出图片
> ggforest(fit.cox)
##二、使用连续变量计算HR
> fit.cox <- coxph(Surv(OS,OS.status) ~ Age,data = plot_data)
> fit.cox.sum <- summary(fit.cox)
> fit.cox.sum$coefficients[2]
[1] 1.013108
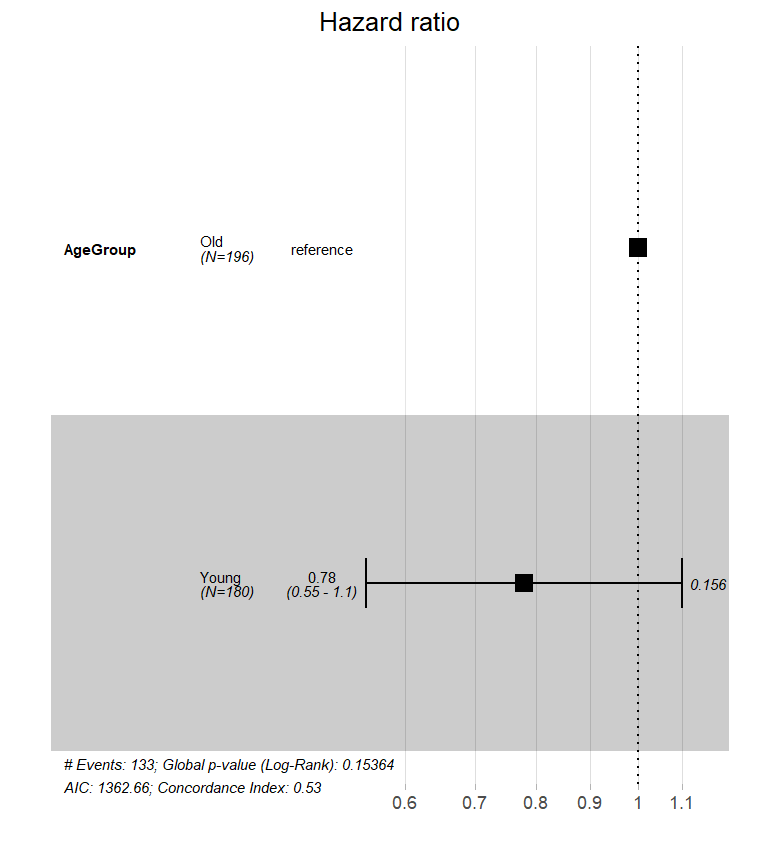
#多因素生存分析(cox计算HR)
fit.cox <- coxph(Surv(OS,OS.status) ~ Age + Gender + Vascular,data = plot_data)
ggforest(fit.cox)生存分析以及预后模型nomogram
- 研究目的:从所有变量中筛选出一组数据生成一个模型来预测患者的死亡风险OS
- 需要用到的包
rms - 预后模型列线图nomogram
#加载包和函数
library(dplyr)
library(survival)
library(survminer)
library(rms)
library(pROC)
library(RColorBrewer)
source("../Codes/Surv_box_plot.R")##读入数据
> nomo_data <- readRDS("./ReadData/nomogrram_test.RDS")
##清洗数据
> nomo_cols <- setdiff(colnames(nomo_data),c("Sample_ID","OS","OS.status"))#取nomo_data所有指标与必用分析指标的差集
> NAvalue <- apply(nomo_data[,nomo_cols],1,function(x)sum(is.na(x)))
> nomo_test <- nomo_data[NAvalue == 0,c(nomo_cols,"SamID","OS","OS.status")]#去除含有NA的样本
> nrow(nomo_test)
[1] 169
> cox.full <- eval(parse(text=paste("coxph(Surv(OS,OS.status)~",paste(nomo_cols,collapse = "+"),",data = nomo_test)",sep="")))
> cox.step <- step(cox.full,direction = "both",trace = T)# 逐步回归的方法筛选重要的因素
> clin_sig <- nomo_test[,c("OS","OS.status","Age","Stage","Tstage","Nstage","iTreg","Th1","ImmuneScore","Monocyte","Cytotoxic")]#将最终筛选出的指标选入新数据
##分析数据(逐步回归法)
# 生成最终的cox预后模型,并基于该模型预测训练集的风险score
model_train = coxph(Surv(OS,OS.status)~Age + Stage + Tstage +
Nstage + iTreg + Th1 + ImmuneScore + Monocyte + Cytotoxic,clin_sig)
clin_sig$p_train = predict(model_train,clin_sig)
# 将风险score按照中位值以及正负值分成高低风险两组
clin_sig <- clin_sig %>%
mutate(Group = ifelse(p_train > 0,"RiskHigh","RiskLow")) %>%
mutate(RiskMedian = ifelse(p_train >= median(p_train),"RiskHigh","RiskLow"))
##输出KM曲线
# 高低风险组的预后对比KM曲线
p <- SurvPlot(plot_data = clin_sig,group_label = "Group",type = "cat",survival = "OS",label.x = 50)
ggsave("./Out/Riskmodel_survival_test.pdf",plot = p$plot,width = 4.5,height = 4)
##输出nomogram模型(列线图)
> dd <- datadist(clin_sig)
> options(datadist="dd")
> f2 <- cph(Surv(OS,OS.status)~Age + Stage + Tstage +
+ Nstage + iTreg + Th1 + ImmuneScore + Monocyte + Cytotoxic,
+ data=clin_sig, x=T, y=T, surv = T, time.inc = 12)
> surv <- Survival(f2)
> nom <- nomogram(f2, fun=list(function(x) surv(24, x),
+ function(x) surv(36, x),
+ function(x) surv(60, x)),
+ funlabel=c("OS > 2Y Probability",
+ "OS > 3Y Probability",
+ "OS > 5Y Probability"))
> pdf("./Out/Nomogram_new_12.pdf",width = 10,height = 8)
> plot(nom, xfrac=.25,cex.axis = 1,cex.var = 1) 

ROC曲线与PCA分析
ROC曲线
理解ROC曲线(受试者特征曲线)之前需要知道两个量——敏感性和特异性
- 敏感性:预测的真阳性占金标准真阳性的百分比(即阳性的正确比例)
特异性:预测的真阴性占金标准真阴性的百分比(即阴性的正确比例)
- 提高敏感性特异性就会降低 ,反之同理
- PPV:预测的真阳性占总数的百分比
- NPV:预测的真阴性占总数的百分比
ROC曲线,本质上就是敏感性和特异性此消彼长的过程,ROC曲线下的面积可以定量地评价模型效果,记作AUC,AUC越大则模型效果越好
平移对角线,与ROC曲线相切,得到AUC
# 生成最终的cox预后模型,并基于该模型预测训练集的风险score
model_train = coxph(Surv(OS,OS.status)~Age + Stage + Tstage +
Nstage + iTreg + Th1 + ImmuneScore + Monocyte + Cytotoxic,clin_sig)
clin_sig$p_train = predict(model_train,clin_sig)
# 基于风险score预测OS死亡
roc1=roc(clin_sig$OS.status,clin_sig$p_train)
roc1$auc
# 画ROC曲线
pdf("./Out/Model_ROC_new.pdf")
plot.roc(clin_sig$OS.status,clin_sig$p_train,
cex.axis=1.2,cex.lab = 1.5,
col = brewer.pal(12,"Set3")[1], print.auc=T)
legend("bottom",
legend = c("Training Set AUC=0.729(N=169,Test)"),
col = brewer.pal(12,"Set3")[1], lwd=2,
cex=1.4, pt.cex = 1, bty = "n")
dev.off()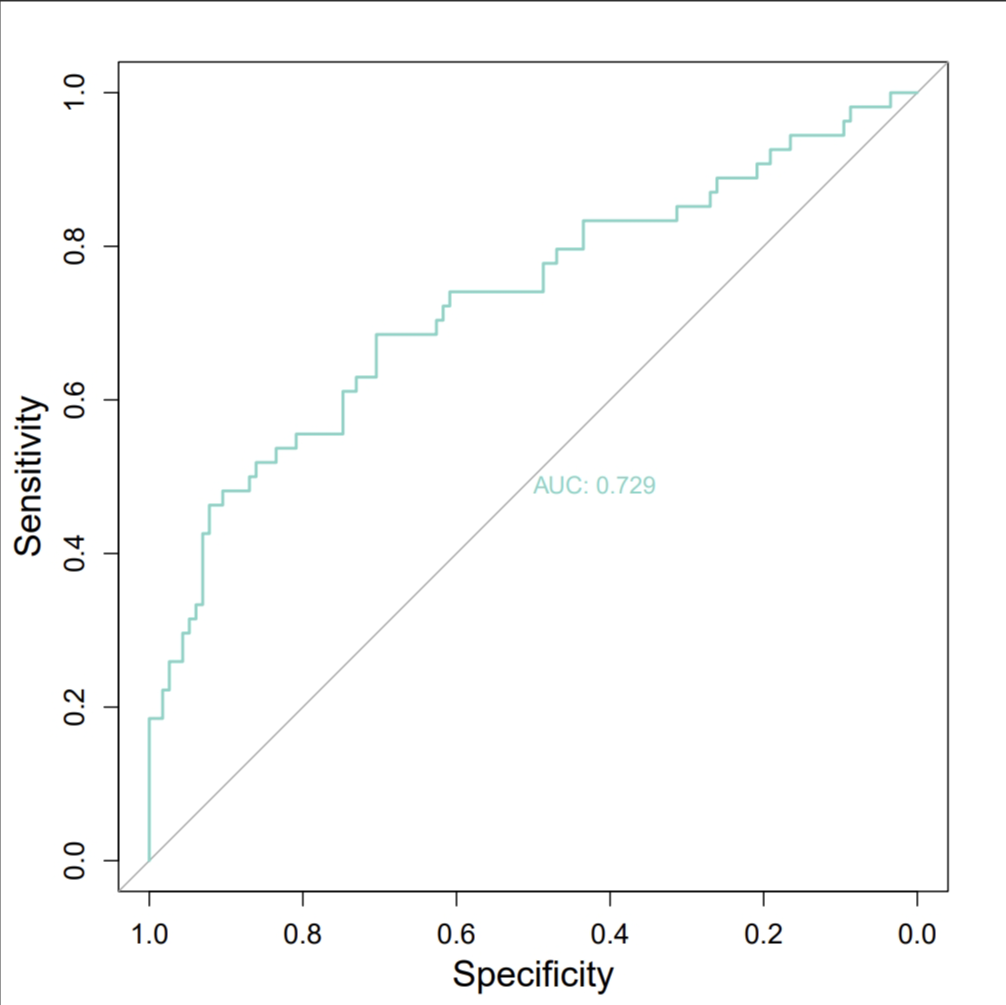
PCA分析
- 降维:是一种对高维度特征数据预处理的方法。降维是将高维度的数据保留下来最重要的一些特征,去除噪声和不重要的特征,从而使实现提升数据处理速度的目的。
- PCA是最广泛的数据降维算法。主要思路是将n维特征映射到k维上。k维是全新的正交特征也被称为主成分,是在原有n维特征的基础上重新构造出来的k维特征。
library(ggbiplot)
# 设置随机数
set.seed(666)
# 筛选肿瘤和对照各50例样本
tcga_clic <- tcga_clic %>%
filter(Sample_ID %in% colnames(tcga_expr))
sam_sel_norm <- sample(tcga_clic$Sample_ID[tcga_clic$SamType == "Normal"],50)
sam_sel_tumor <- sample(tcga_clic$Sample_ID[tcga_clic$SamType == "Primary"],50)
# 合并成100样本矩阵并进行Z-score转换,去除含有NA的基因
plot_pct <- tcga_expr[,c(sam_sel_norm,sam_sel_tumor)]
plot_pct <- t(scale(t(plot_pct)))
NAgenes <- apply(plot_pct,1,function(x)ifelse(sum(is.na(x)),T,F))
plot_pct <- plot_pct[!NAgenes,]
# 筛选500个基因进行PCA分析测试:列为基因,行为样本,需要转置
pr_test <- prcomp(t(plot_pct[1:500,]))
ggbiplot(pr_test,var.axes = FALSE,
groups = c(rep("Normal",50),rep("Tumor",50)))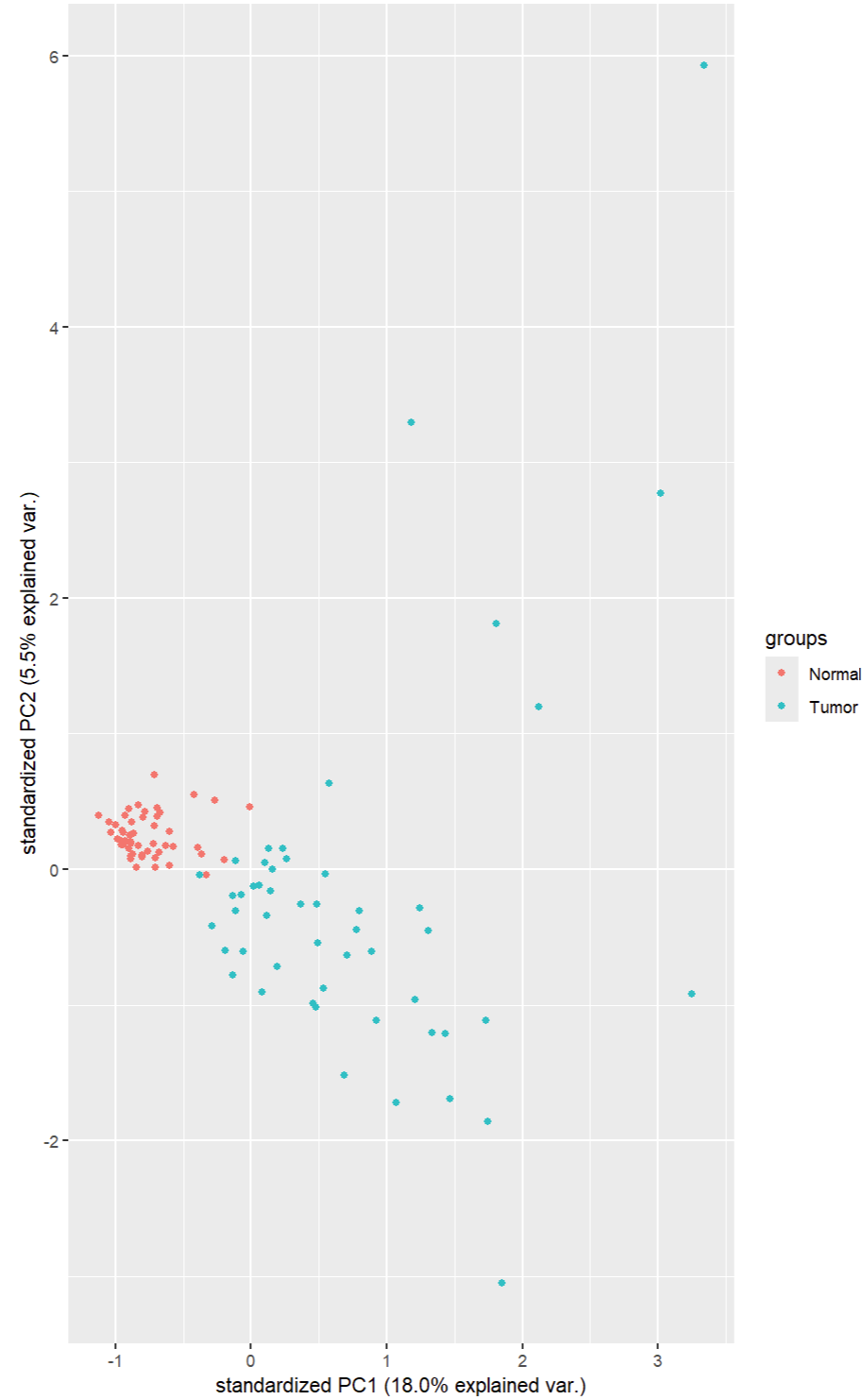
六、R语言临床(思路)
- 统计分析中的变量或者关注因素
- 用R语言批量完成统计检验筛选显著相关变量
统计背后的两个核心方向
- 分析差异是否显著
- 分析相关性是否显著
不同类型变量之间的比较方法
- 连续vs连续——cor.test
- 连续vs分类——wilcoxon test
- 分类vs分类——chisq or fisher test
- 分类vs预后——log-rank
- 连续vs预后——coxph
自动化统计相关性(要研究哪些)
清洗数据
(感觉讲得有点晚了,应该早点说)
要点:
- 数值类型中全是数值,不是的变为NA
- 分类类型中类别不能太多,适当合并或去除
- 预后类型用石油PFS与PFS.status,OS,DFS同理
- 每行是一个样本,每列是一个变量
定义变量
5组变量
- gene_con
- gene_cat
- clinical_con
- clinical_cat
- survival

具体可见“三、R函数学习1——一些数据挖掘中的基础统计分析——批量进行分析统计”
WGCNA分析(加权共表达网络分析)
概念:WGCNA是用来描述不同样品之间基因关联模式的系统生物学办法,可以用来鉴定高度协同变化的基因集,并根据基因集的内连性和基因集与表型之间的关联鉴定候补生物标记基因或治疗靶点。
WGCNA在数据挖掘中常被用于从数量较多的基因中寻找到其中的关键基因或者关键基因组合
WGCNA分析步骤
- 筛选基因创建共表达网络(常见筛选基因的方法包括差异表达基因,TOP-MAD基因)
- 构建共表达网络计算其中的基因modules
- 计算gene module与其他临床因素的相关性
- 从module中筛选得到的核心基因
实例分析
##数据读入
load("./ReadData/datExpr.rdata") # 筛选的5000个基因
load("./ReadData/datTraits.rdata") # 整理之后的临床数据
# 自定义一个样本聚类函数用于后续寻找离群样本
sampleTree = function(datExpr) {
nGenes = ncol(datExpr)
nSamples = nrow(datExpr)
datExpr_tree <- hclust(dist(datExpr), method = "average")
par(mar = c(0, 5, 2, 0))
plot(
datExpr_tree,
main = "Sample clustering",
sub = "",
xlab = "",
cex.lab = 2,
cex.axis = 1,
cex.main = 1,
cex.lab = 1
)
datExpr_tree
}##清洗数据(剔除离群样本)
# 先画出全部样本的聚类树观察是否有比较明显的离群样本
sampleTree(datExpr)
# 假如有则将聚类树存到一个变量中,筛选一个cutoff把图中的离群样本删除
datExpr_tree = sampleTree(datExpr)
height_cut = 350 #筛选离群样本的阈值需要结合图以及分析的结果定
clust = cutreeStatic(datExpr_tree, cutHeight = height_cut, minSize = 10)
table(clust)
keepSamples = (clust==1)
dim(datExpr)
datExpr = datExpr[keepSamples, ]
dim(datExpr)
# 观察结果
sampleTree(datExpr)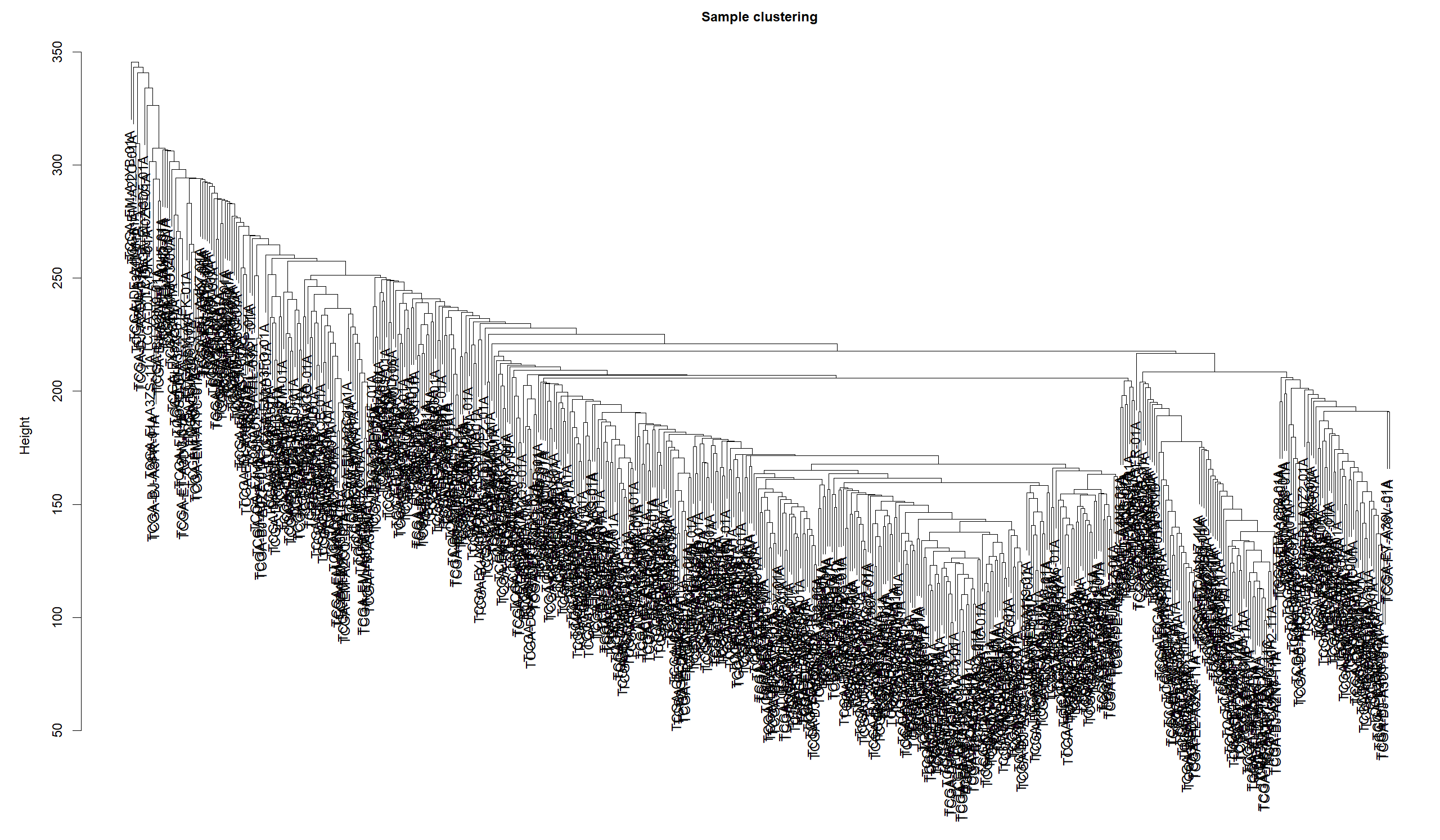
##确定最佳BETA值
powers = c(c(1:10), seq(from = 12, to = 20, by = 2))
sft = pickSoftThreshold(datExpr, powerVector = powers, verbose = 5)
pdf("./WGCNA/1_beta.pdf",width = 8,height = 5)
par(mfrow = c(1, 2))
cex1 = 0.9
plot(
sft$fitIndices[, 1],
-sign(sft$fitIndices[, 3]) * sft$fitIndices[, 2],
xlab = "Soft Threshold (power)",
ylab = "Scale Free Topology Model Fit,signed R^2",
type = "n",
main = paste("Scale independence")
)
text(
sft$fitIndices[, 1],
-sign(sft$fitIndices[, 3]) * sft$fitIndices[, 2],
labels = powers,
cex = cex1,
col = "red"
)
abline(h = 0.90, col = "red")
plot(
sft$fitIndices[, 1],
sft$fitIndices[, 5],
xlab = "Soft Threshold (power)",
ylab = "Mean Connectivity",
type = "n",
main = paste("Mean connectivity")
)
text(
sft$fitIndices[, 1],
sft$fitIndices[, 5],
labels = powers,
cex = cex1,
col = "red"
)##构建共表达矩阵
net = blockwiseModules(
datExpr,
power = sft$powerEstimate,
maxBlockSize = 6000,
TOMType = "unsigned",
minModuleSize = 30,
reassignThreshold = 0,
mergeCutHeight = 0.25,
numericLabels = TRUE,
pamRespectsDendro = FALSE,
saveTOMs = TRUE,
saveTOMFileBase = "TEST",
verbose = 3
)##模块可视化
table(net$colors)
# Convert labels to colors for plotting
mergedColors = labels2colors(net$colors)
write.xlsx(table(mergedColors),file = "./WGCNA/2_mergedColors_count.xlsx")
# Plot the dendrogram and the module colors underneath
pdf("./WGCNA/2_modules.pdf",width = 6,height = 5)
plotDendroAndColors(
net$dendrograms[[1]],
mergedColors[net$blockGenes[[1]]],
"Module colors",
dendroLabels = FALSE,
hang = 0.03,
addGuide = TRUE,
guideHang = 0.05
)
dev.off()六、R语言实践
回顾
- R语言基础:数据类型、数据结构、数据清洗
- 代码进阶:常用函数整理,自建代码库,理解并修改代码
- 医学领域常用:ggplot2画图,热图聚类分析,生存分析,WGCNA分析等
数据的下载与清洗
下载
清洗整理
整理过程
- 去掉低表达量的基因
- 去掉表达量几乎一致的基因
- 去掉没有临床信息或者OS<30天的样本
- 去掉癌旁对照样本
- TCGA的数据集随机分为两组,一组作为训练集,,一组作为验证集
- pathway的基因经过筛选之后得到594个
分析
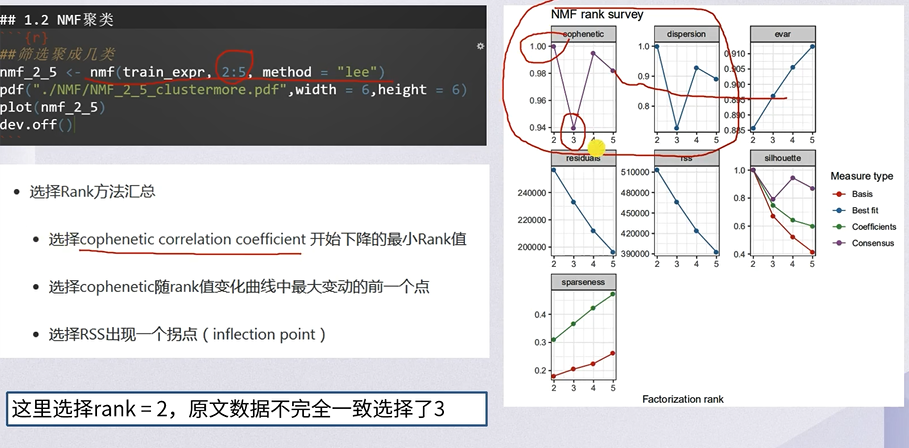
第一步:样本聚类
方法一:通过Complexheatmap进行层级聚类
方法二:NMF聚类法
第二步:生存分析,差异分析,富集分析
补充:差异分析补充-DESeq2使用
第三步:构建预后模型
- 先通过coxph计算每一个基因的预后显著性,然后按照p<0.01的标准把基因筛出来
- 将筛选出来的基因用lasso-cox模型/逐步回归方法筛选最优模型
- 模型ROC评估以及预后评估
第四步:独立数据集验证
第五步:实验验证基因的表达特征
数据挖掘的方法
POLE和POLD1突变作为多种癌症类型免疫治疗结果生物标志物的评估
中高级数据挖掘文章的规划思路
- 研究选题:回答什么问题(重要性)?创新性?
- 方案设计:如何实现?结论是否可靠?
第一步:选题
idea的来源:抄概念
- What is research question?
- Why is it important?
- What is the rational of investigating the exposure/intervention and outcome?
- Why do we need a new study?
第二步:研究设计
实例说明
- 研究目的:免疫联合治疗vs免疫单药治疗,在二线及以上NSCLC患者中的疗效以及安全性
研究设计:PECOS
- P(研究人群): 接受免疫治疗或免疫联合治疗作为二线或多线治疗的晚期NSCLC患者
- E(干扰因素): PD-1联合化疗;联合抗血管生成;联合化疗及抗血管生成
- C(研究因素): PD-1单药治疗
- O(核心目的): PFS; secondary ORR, OS, safety
- S(研究类型): 非随机对照研究(回顾性队列研究)
- 研究混杂因素控制:治疗线数、上一线治疗方案、病理类型、转移部位、转移灶数量、PD-1使用时间,不同PD-1,联合的方案,PS评分,4周内放疗史,炎症,抗生素种类,LDH






3 条评论
我勒个痘痘痘
捉!高仿!
一下子撸了这么多,牛逼